TCMNP包教程:这个R包够猛!除了药物+疾病+靶点+转录因子数据库,还能做网络药理学全流程可视化分析?!
生信碱移TCMNP 包,一路打通网络药理学数据库与可视化分析。最近,小编冲浪的时候看到一个今年发表的网络药理学 R 包 TCMNP,据说整合了多个数据库以及多种可视化分析函数,一眼看下来确实够顶。具体来说,TCMNP 整合了几大类型的数据库:① 三大药物成分及靶点数据库TCMSP、DrugBank、ETCM;② 疾病基因数据库 DisGeNET 与 OMIM;③ 转录因子及其对应靶基因数据库 TR
生信碱移
TCMNP包
TCMNP 包,一路打通网络药理学数据库与可视化分析。
最近,小编冲浪的时候看到一个今年发表的网络药理学 R 包 TCMNP,据说整合了多个数据库以及多种可视化分析函数,一眼看下来确实够顶。

DOI: 10.1016/j.prmcm.2024.100562。
具体来说,TCMNP 整合了几大类型的数据库:① 三大药物成分及靶点数据库TCMSP、DrugBank、ETCM ;② 疾病基因数据库 DisGeNET 与 OMIM;③ 转录因子及其对应靶基因数据库 TRRUST。这几个数据库涵盖了 571 种中药材、17118 种化合物、10013 种疾病和 15956 个靶点,可以都是小编认知中热门的几个数据库了。基于这些数据,TCMNP 自行自动化的网络药理学分析与可视化,具体可以直接看下图:

图:TCMNP 九大功能。1.与中药相关的内容,包括药材、化合物和靶点;2.疾病信息,包括疾病及其相关靶基因;3.中药化合物成分的可视化;4.京都基因与基因组百科全书(KEGG)和基因本体(GO)富集分析;5.基因集富集分析(GSEA);6.蛋白质相互作用(PPI)网络分析;7.转录因子筛选与可视化;8.药物与疾病相互作用分析;9.分子对接评分后的热图可视化。
看到这小编想说,网络药理学的常规分析也就这些了吧。而且通过这个R包整合的数据和函数,进行一些高级分析也十分快速。这里咱就简要介绍该包主要使用函数,对具体的处理参数以及结果描述,建议阅读下方链接以及原文:
-
https://github.com/tcmlab/TCMNP
-
数据库:https://tcmlab.com.cn/tcmnp/
0.R包安装与加载
使用下方代码安装该R包:
if(!require(devtools))install.packages("devtools")
if(!require(TCMNP))devtools::install_github("tcmlab/TCMNP",upgrade = FALSE,dependencies = TRUE)
加载一些需要的R包:
library(TCMNP)
library(dplyr)
library(ggplot2)
library(ggraph)
library(clusterProfiler, quietly= TRUE)
library(org.Hs.eg.db, quietly= TRUE)
library(DOSE, quietly= TRUE)
TCMNP使用示例
① 可视化药物组成:
xfbdf.compostion <- data.frame(
herb = c(
"mahuang", "kuxingren", "shengshigao",
"shengyiren", "maocangzhu", "guanghuoxiang",
"qinghaocao", "mabiancao", "ganlugen", "tinglizi",
"huajuhong", "shenggancao", "huzhang"
),
weight = c(6, 15, 30, 30, 10, 15, 12, 30, 30, 15, 15, 10, 20)
)
t
cm_comp(xfbdf.compostion)
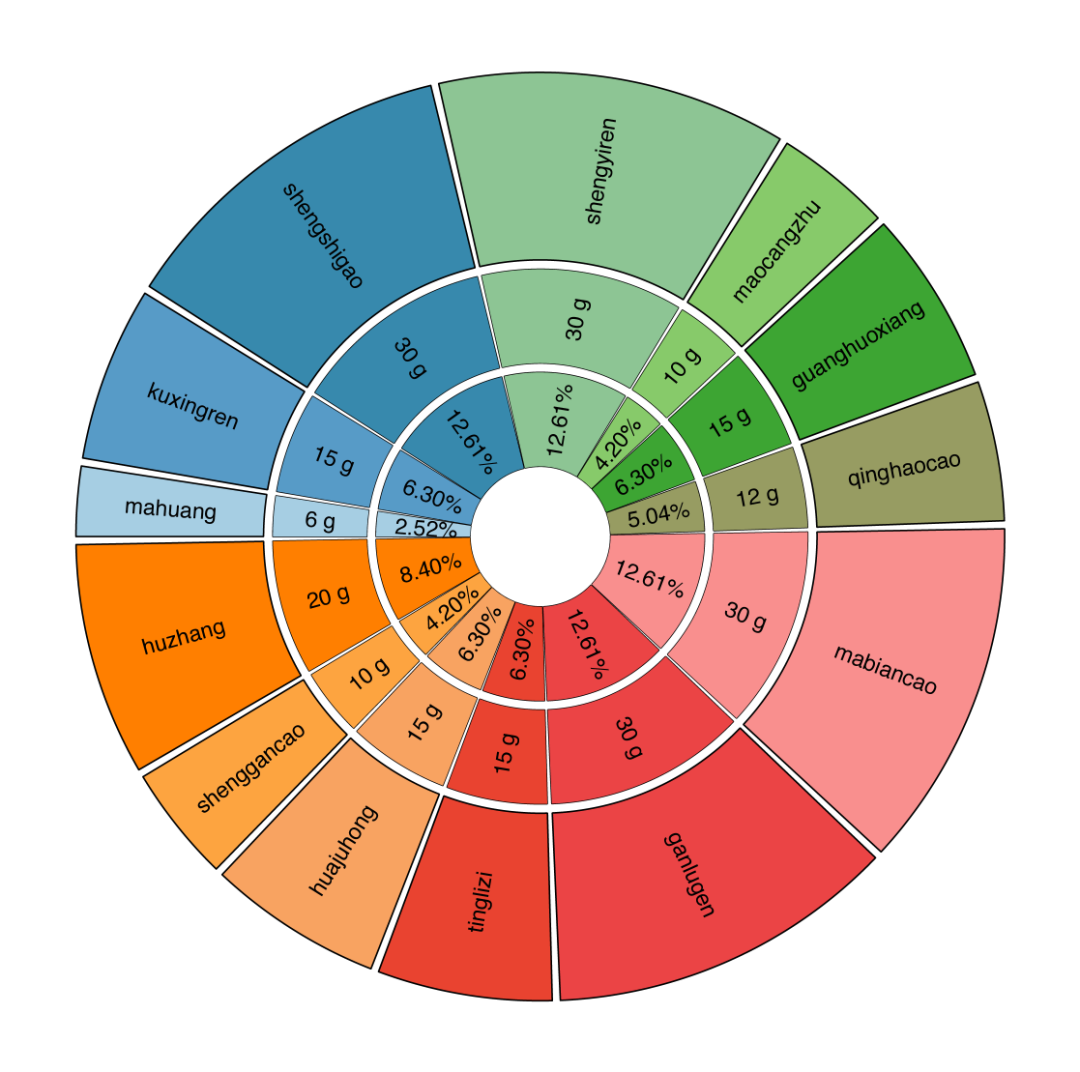
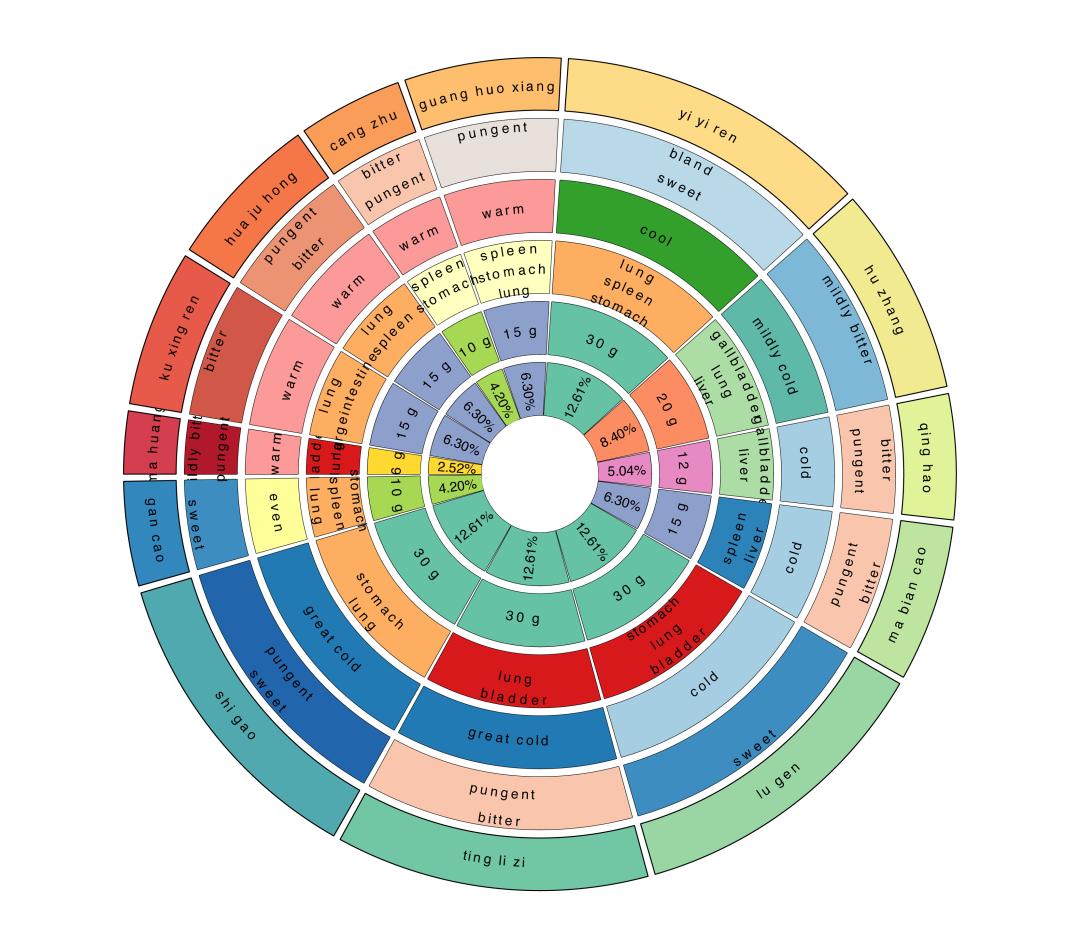
可视化更多信息:
herb = c("ma huang", "ku xing ren", "hua ju hong",
"cang zhu", "guang huo xiang", "yi yi ren",
"hu zhang", "qing hao", "ma bian cao", "lu gen",
"ting li zi", "shi gao", "gan cao")
xfbdf.compostion <- data.frame(
herb = herb,
weight = c(6, 15, 15, 10, 15, 30, 20, 12, 30, 30, 15, 30, 10),
property = herb_pm[match(herb, herb_pm$Herb_name_pinyin), ]$Property,
flavor = herb_pm[match(herb, herb_pm$Herb_name_pinyin), ]$Flavor,
meridian = herb_pm[match(herb, herb_pm$Herb_name_pinyin), ]$Meridian
)
tcm_compound(xfbdf.compostion)
② 获取药物组成以及相应靶点,支持中文或拼音检索:
# 中文检索
herbs<-c('麻黄', '甘草','苦杏仁','石膏',
'薏苡仁', '苍术', '青蒿', '猪苓',
'马鞭草', '葶苈子','化橘红',
'虎杖', '广藿香','芦根')
xfbdf <- herb_target(herbs, type = "Herb_cn_name")
head(xfbdf)
#herb molecule_id molecule target
#cang zhu MOL000173 wogonin NOS2
#cang zhu MOL000173 wogonin PTGS1
#cang zhu MOL000173 wogonin ESR1
#cang zhu MOL000173 wogonin AR
#cang zhu MOL000173 wogonin SCN5A
#cang zhu MOL000173 wogonin PPARG
# 拼音检索
herbs2 <- c("ma huang", "ku xing ren")
fufang2 <- herb_target(herbs2, type ="Herb_name_pin_yin")
head(fufang2)
#herb molecule_id molecule target
#ku xing ren MOL010921 estrone D1R
#ku xing ren MOL010921 estrone DRD1
#ku xing ren MOL010921 estrone CHRM3
#ku xing ren MOL010921 estrone F2
#ku xing ren MOL010921 estrone CHRM1
#ku xing ren MOL010921 estrone CHRM5
③ 靶点反查潜在治疗药物成分以及对应中药(这个功能简单但是实用啊):
gene <- c("MAPK1", "JUN", "FOS", "RAC1", "IL1", "IL6")
herb.data <- target_herb(gene)
head(herb.data)
# herb molecule_id molecule target
#ai di cha MOL000422 kaempferol JUN
#ai di cha MOL000098 quercetin FOS
#ai di cha MOL000098 quercetin MAPK1
#ai di cha MOL000098 quercetin JUN
#ai di cha MOL000098 quercetin IL6
# ai ye MOL000358 beta-sitosterol JUN
④ 通过疾病靶点基因寻找中药及其处方:
# 中药如下
tcm_prescription(disease_data)[[1]]
# A tibble: 273 × 2
# Groups: Herb_cn_name [273]
# Herb_cn_name freq
# <chr> <int>
# 1 人参 437
# 2 甘草 433
# 3 胡芦巴 325
# 4 皂角刺 288
# 5 地榆 276
# ℹ 263 more rows
# ℹ Use `print(n = ...)` to see more rows
# 查看寻找到的处方,截图如下
library(formattable)
formattable(tcm_prescription(disease_data)[[2]], list(Count = color_bar("lightblue")))
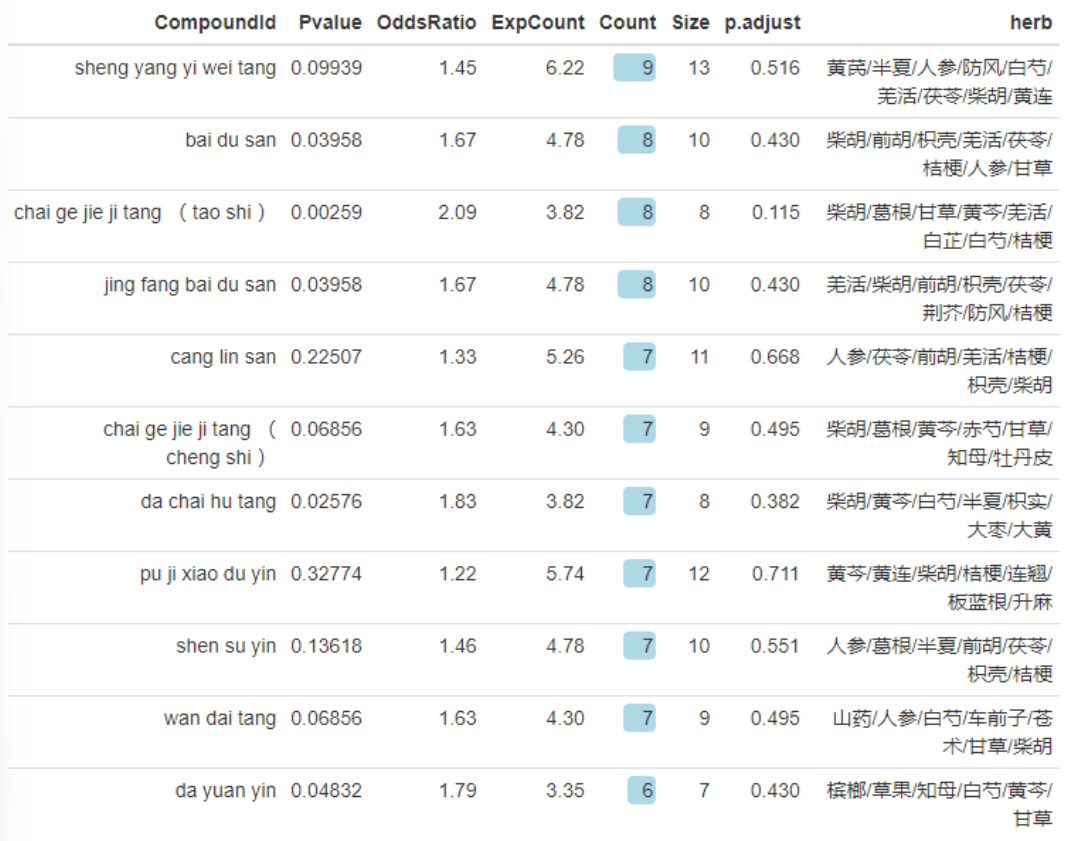
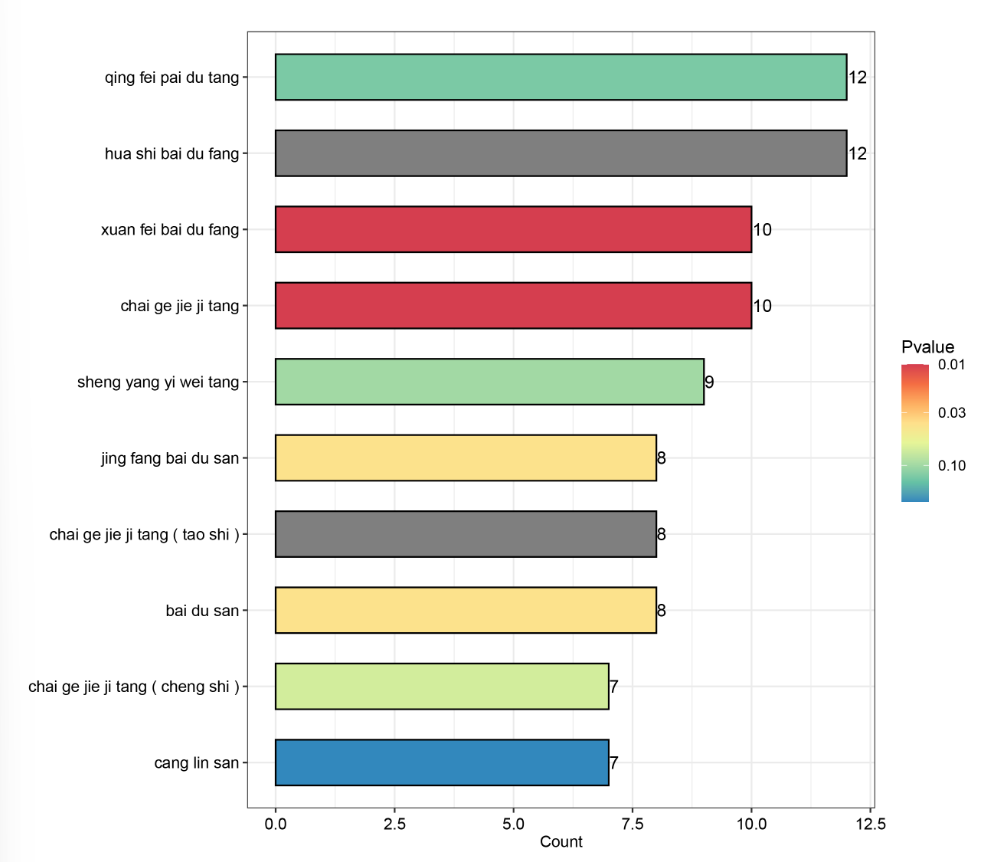
中药处方也来富集?这是小编没想到的,有点surprising。可视化一下:
tcm_pre_plot(tcm_prescription(disease_data)[[2]],color = 'Spectral')
⑤ 药物成分靶点网络:
data("xfbdf", package = "TCMNP")
network.data <- xfbdf %>%
dplyr::select(herb, molecule, target) %>%
sample_n(100, replace = FALSE) %>%
as.data.frame()
tcm_net(network.data,
label.degree = 3,
rem.dis.inter = TRUE)
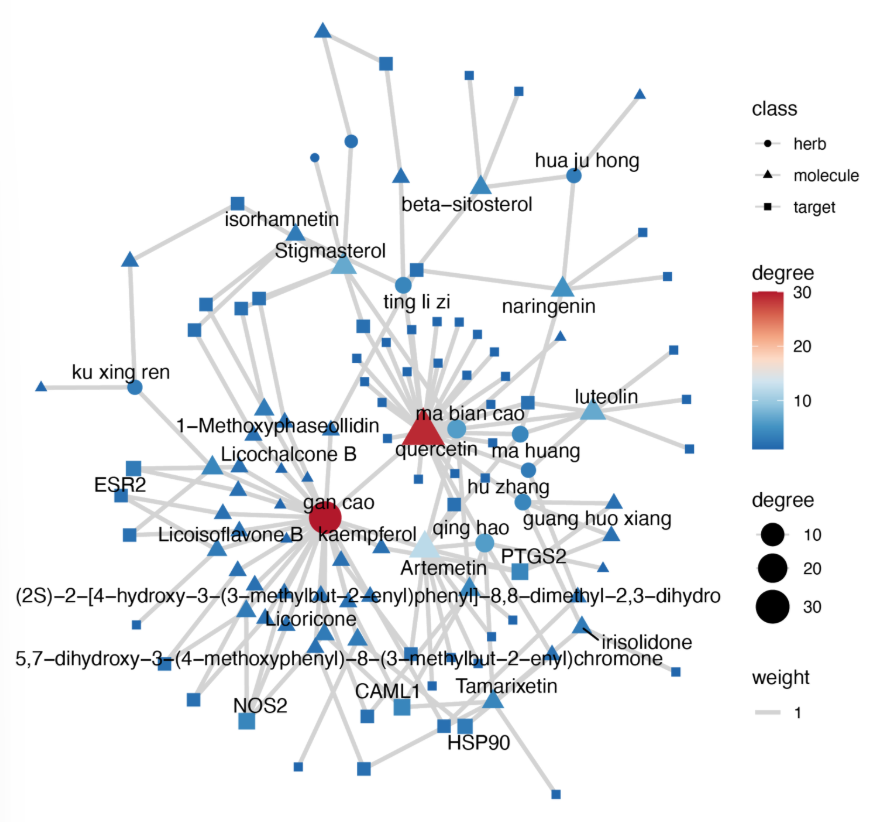
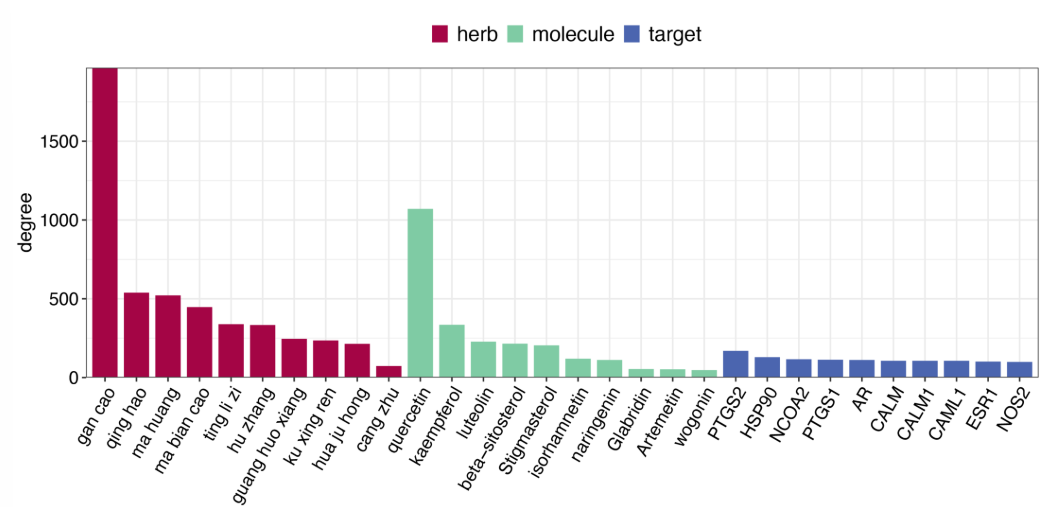
⑥ 可视化药物成分靶点网络的Degree得分:
degree_plot(xfbdf,plot.set='horizontal')
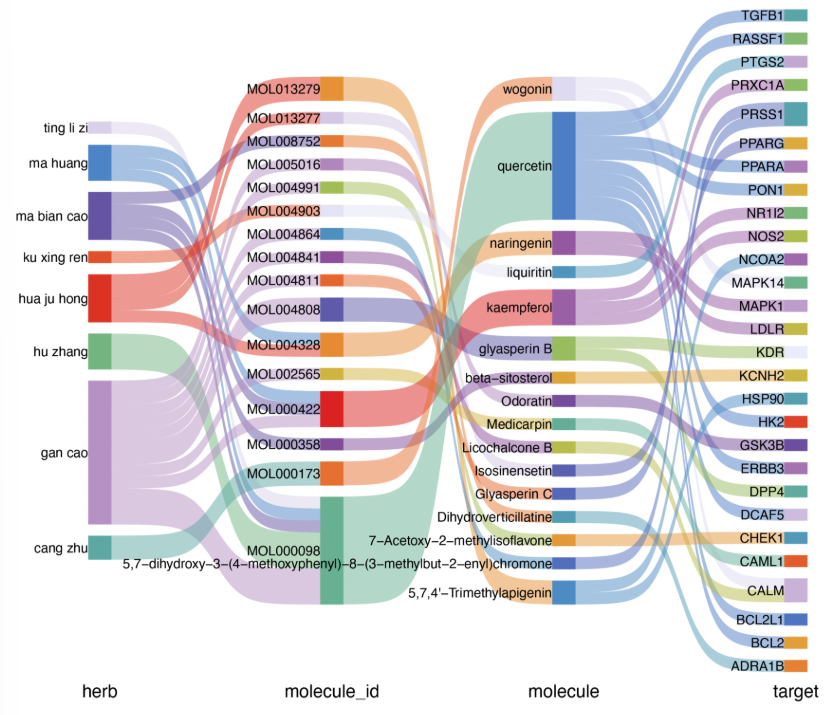
⑦ 药物成分靶点网络桑基图:
sankey.data <- xfbdf[sample(nrow(xfbdf), 30), ]
tcm_sankey(sankey.data,
text.size = 3,
text.position = 1
)
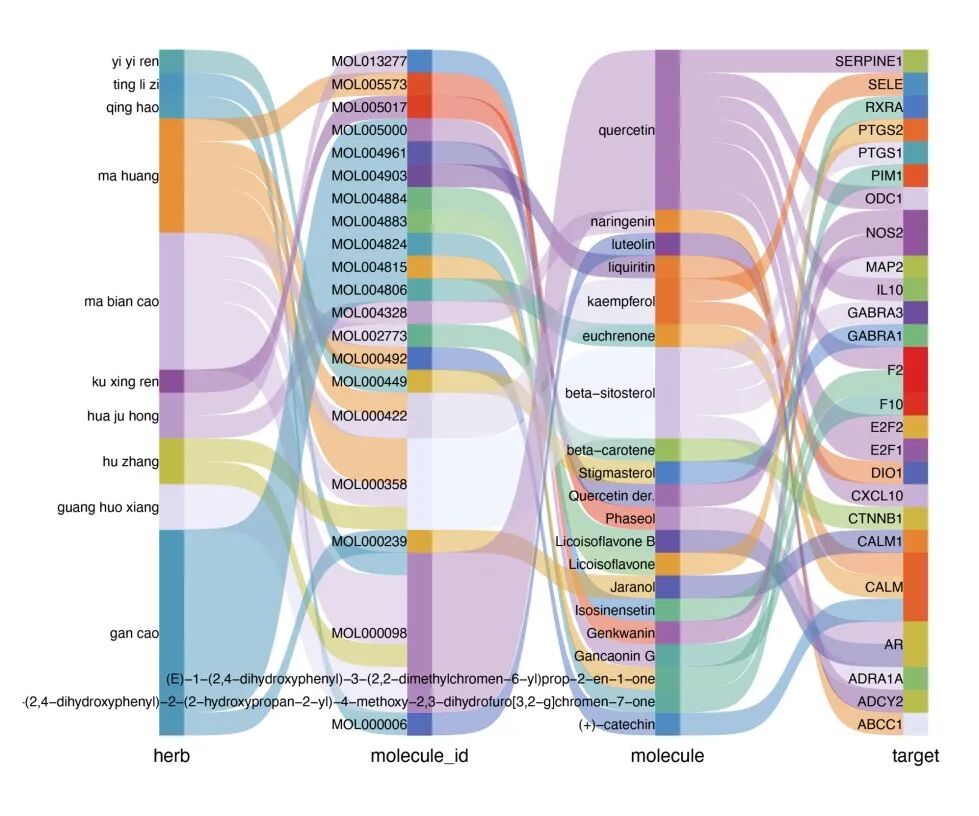
还有冲积图:
alluvial.data <- xfbdf[sample(nrow(xfbdf), 30), ]
tcm_alluvial(alluvial.data,
text.size = 3,
text.position = 1
)
⑧ 药物靶点富集图,首先是KEGG:
data(xfbdf, package = "TCMNP")
eg <- bitr(unique(xfbdf$target), fromType = "SYMBOL", toType = "ENTREZID", OrgDb = "org.Hs.eg.db")
KK <- enrichKEGG(
gene = eg$ENTREZID,
organism = "hsa",
pvalueCutoff = 0.05
)
KK <- setReadable(KK, "org.Hs.eg.db", keyType = "ENTREZID")
bar_plot(KK,title = "KEGG")
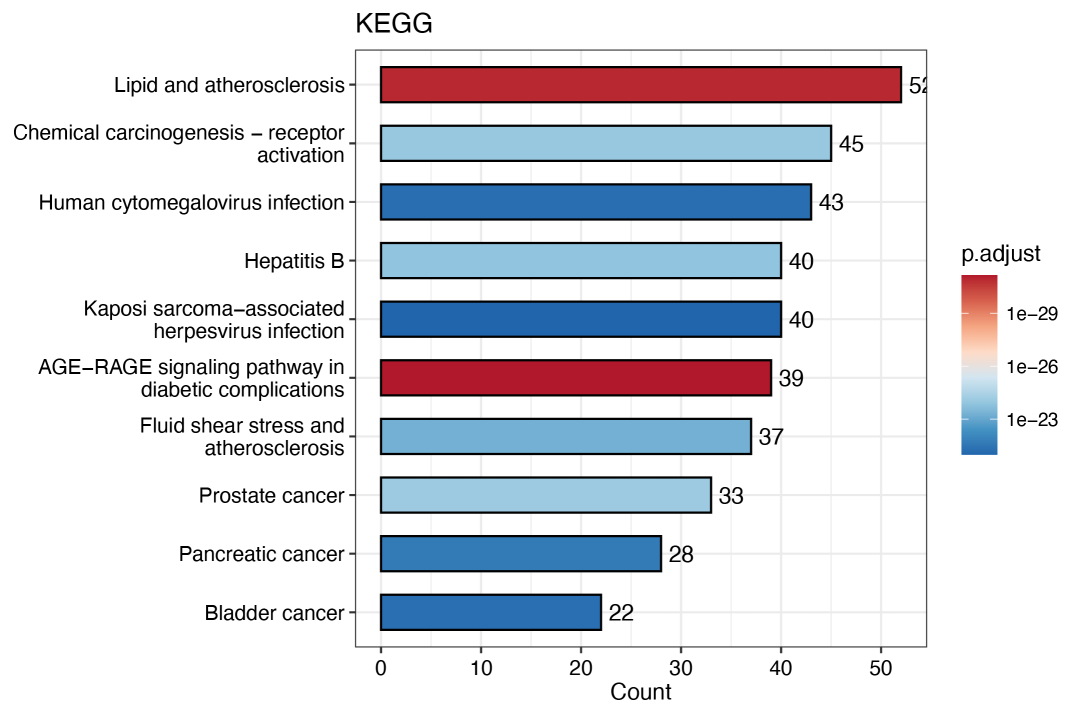
还有GO富集分析:
BP <- enrichGO(
gene = eg$ENTREZID,
"org.Hs.eg.db",
ont = "BP",
pvalueCutoff = 0.05,
readable = TRUE
)
bar_plot(BP,title = "biological process")
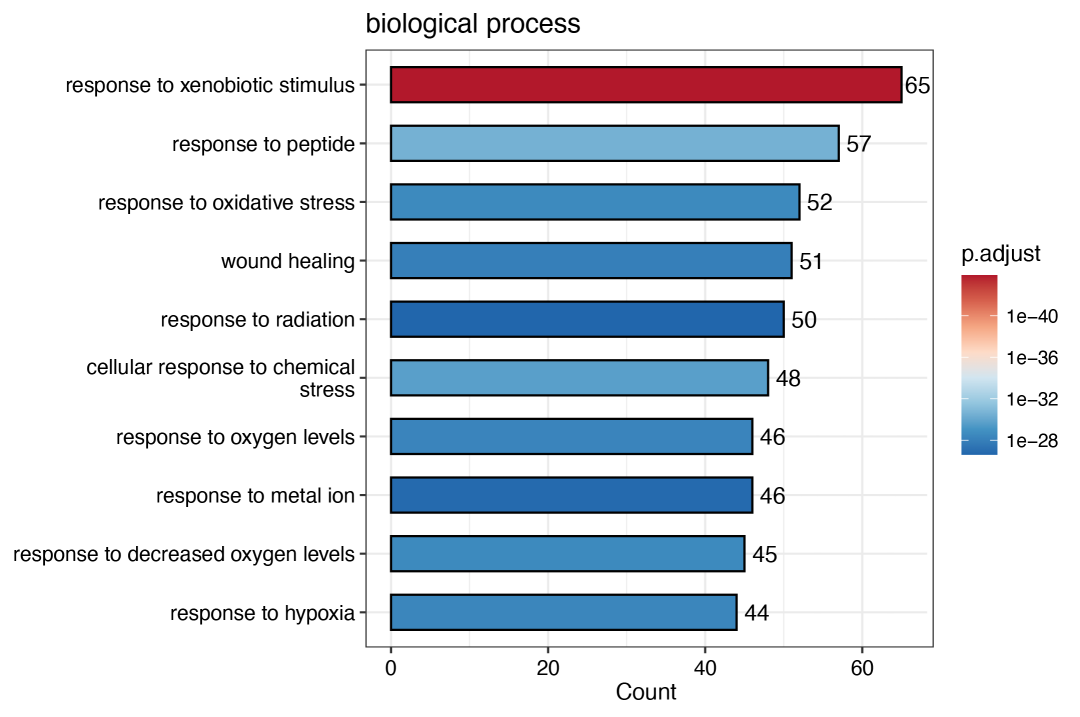
当然可以分面:
eg <- bitr(unique(xfbdf$target), fromType = "SYMBOL", toType = "ENTREZID", OrgDb = "org.Hs.eg.db")
go.diff <- enrichGO(
gene = eg$ENTREZID,
org.Hs.eg.db,
pAdjustMethod = "BH",
pvalueCutoff = 0.01,
qvalueCutoff = 0.05,
ont = "all",
readable = T
)
go_barplot(go.diff)
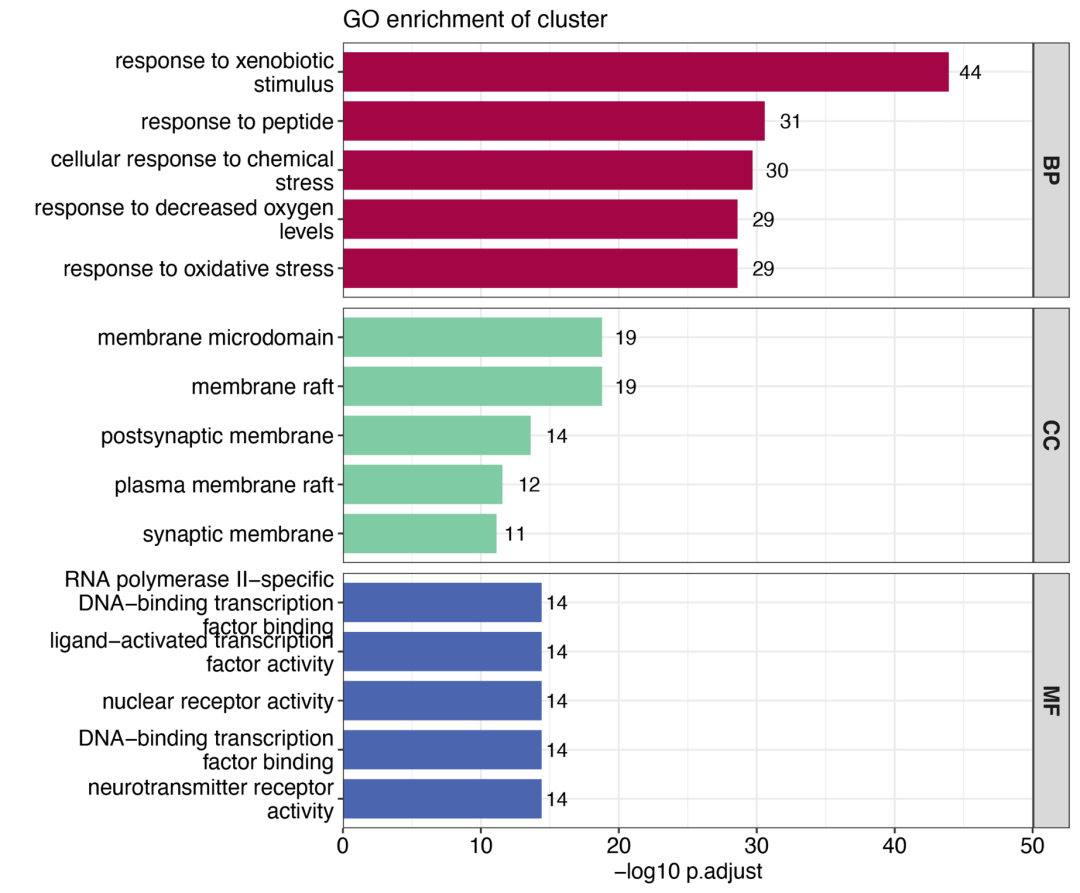
还有点图、棒棒糖图、圈图:
dot_plot(KK, title = "KEGG")
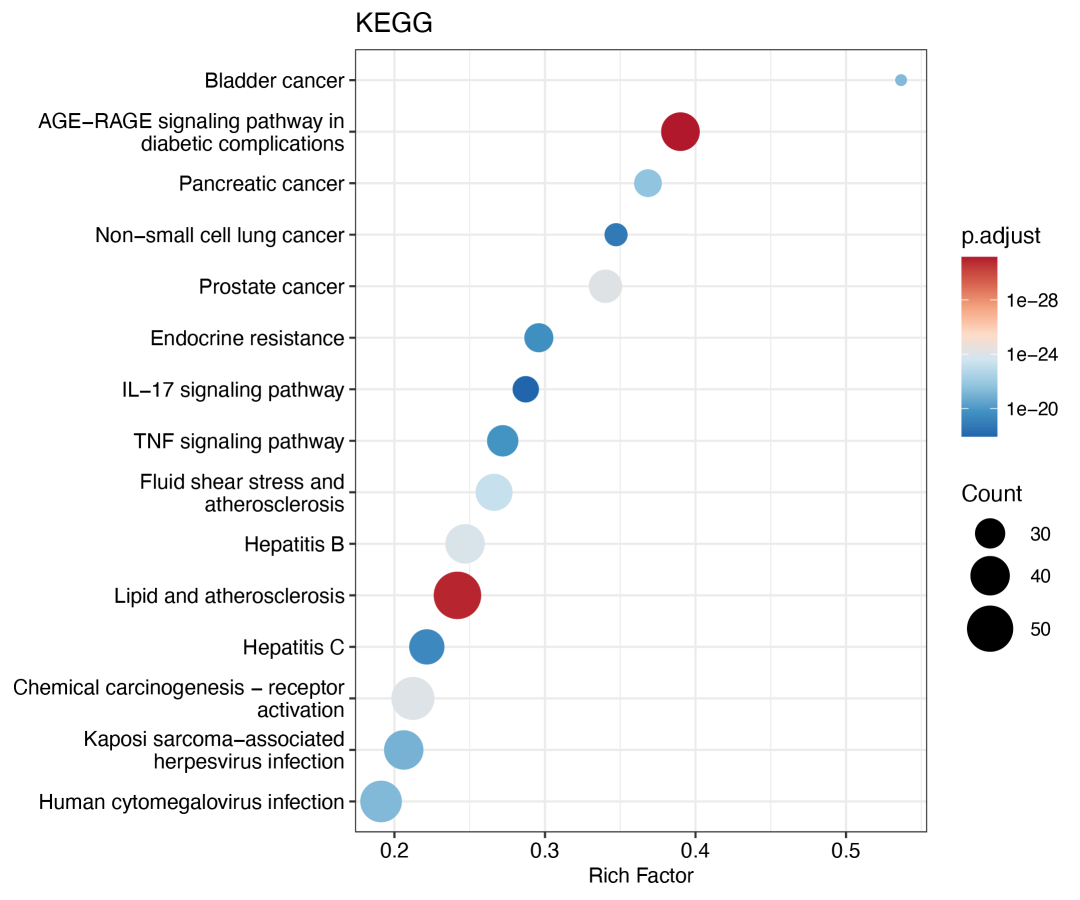
lollipop_plot(KK, title = "KEGG")
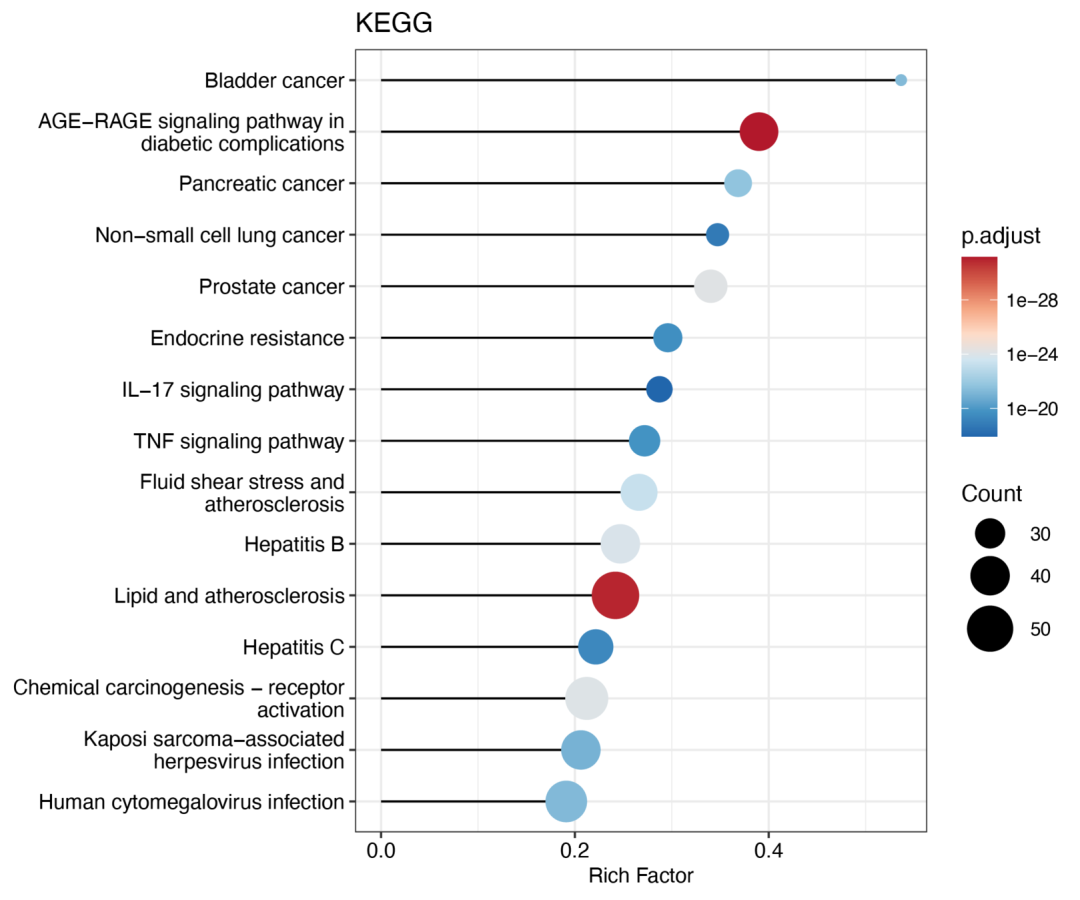
cir_plot(KK)
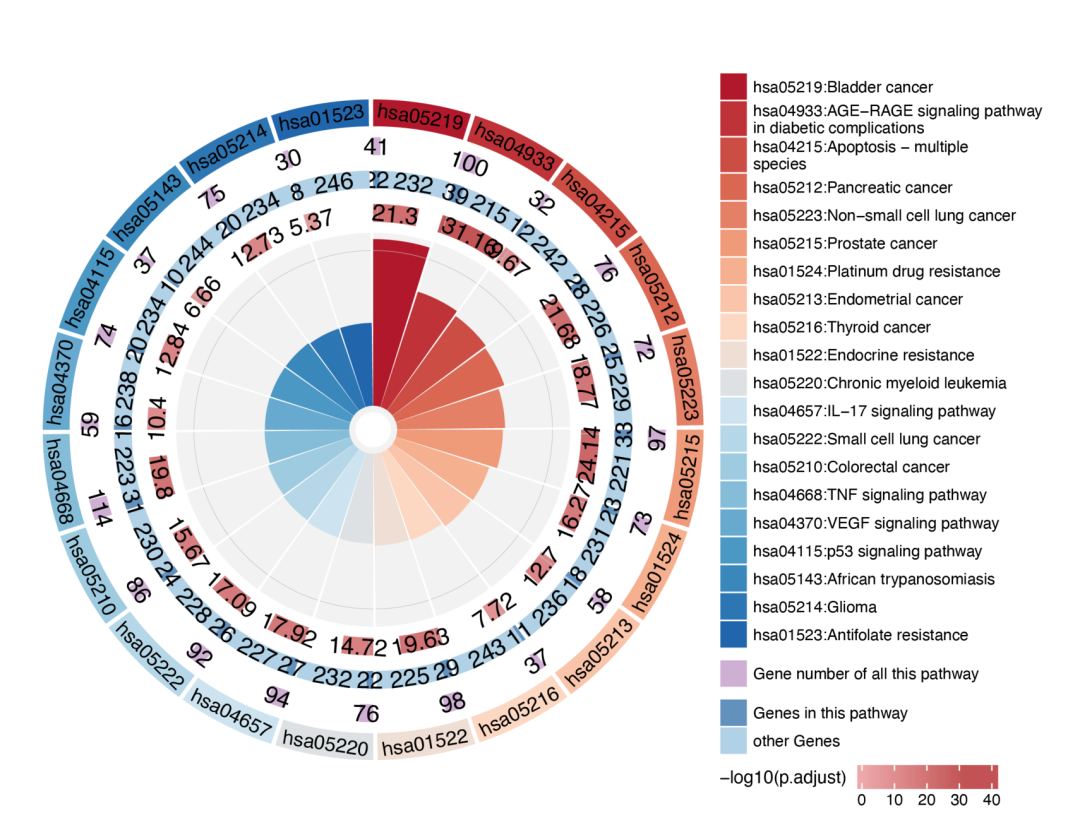
⑨ 基因通路的富集图:
#Filter the pathways to display
data(KK, package = "TCMNP")
newdata <- KK %>% dplyr::slice(11, 15, 17, 33, 53, .preserve = .preserve)
pathway_cirplot(newdata)
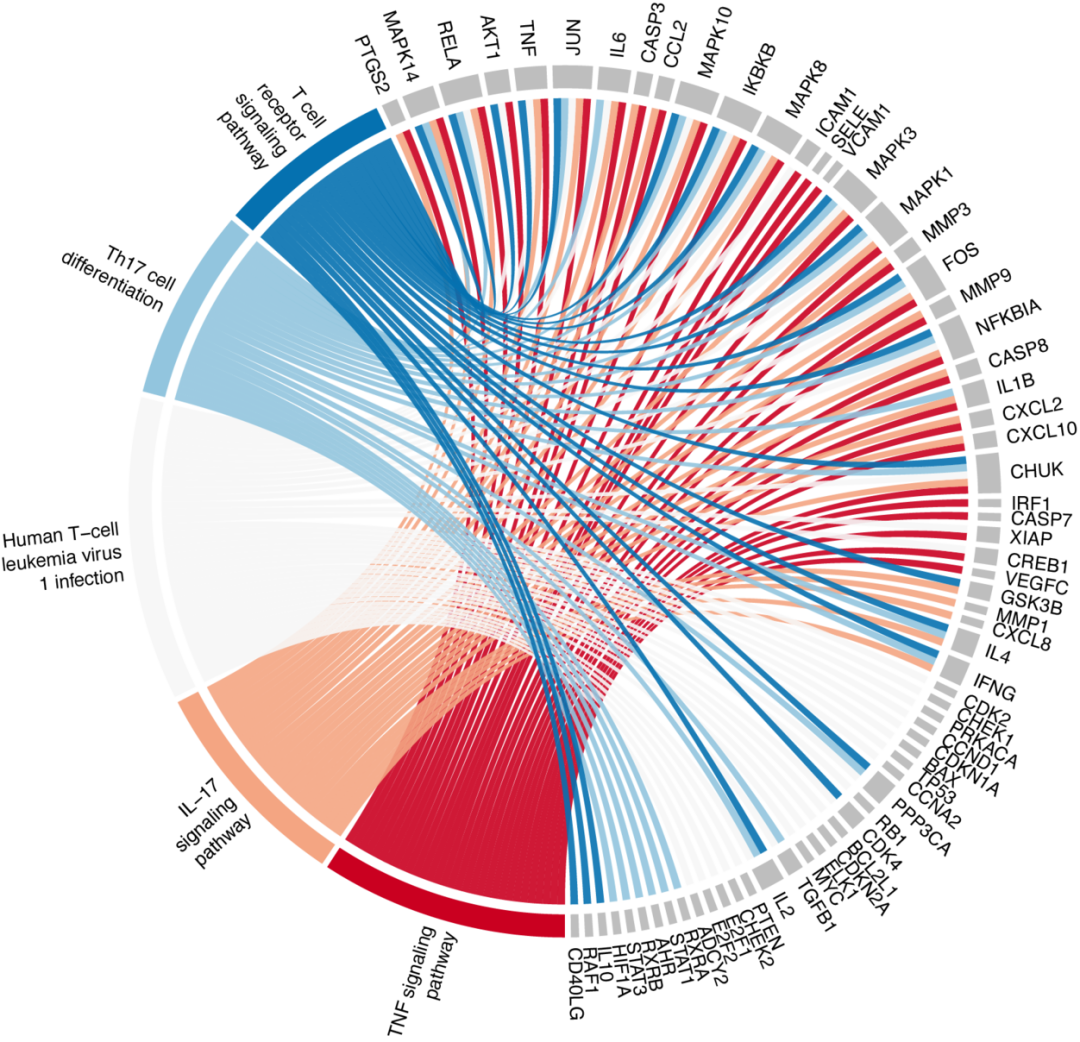
这版更好看:
pathway_ccplot(KK, root = "KEGG")
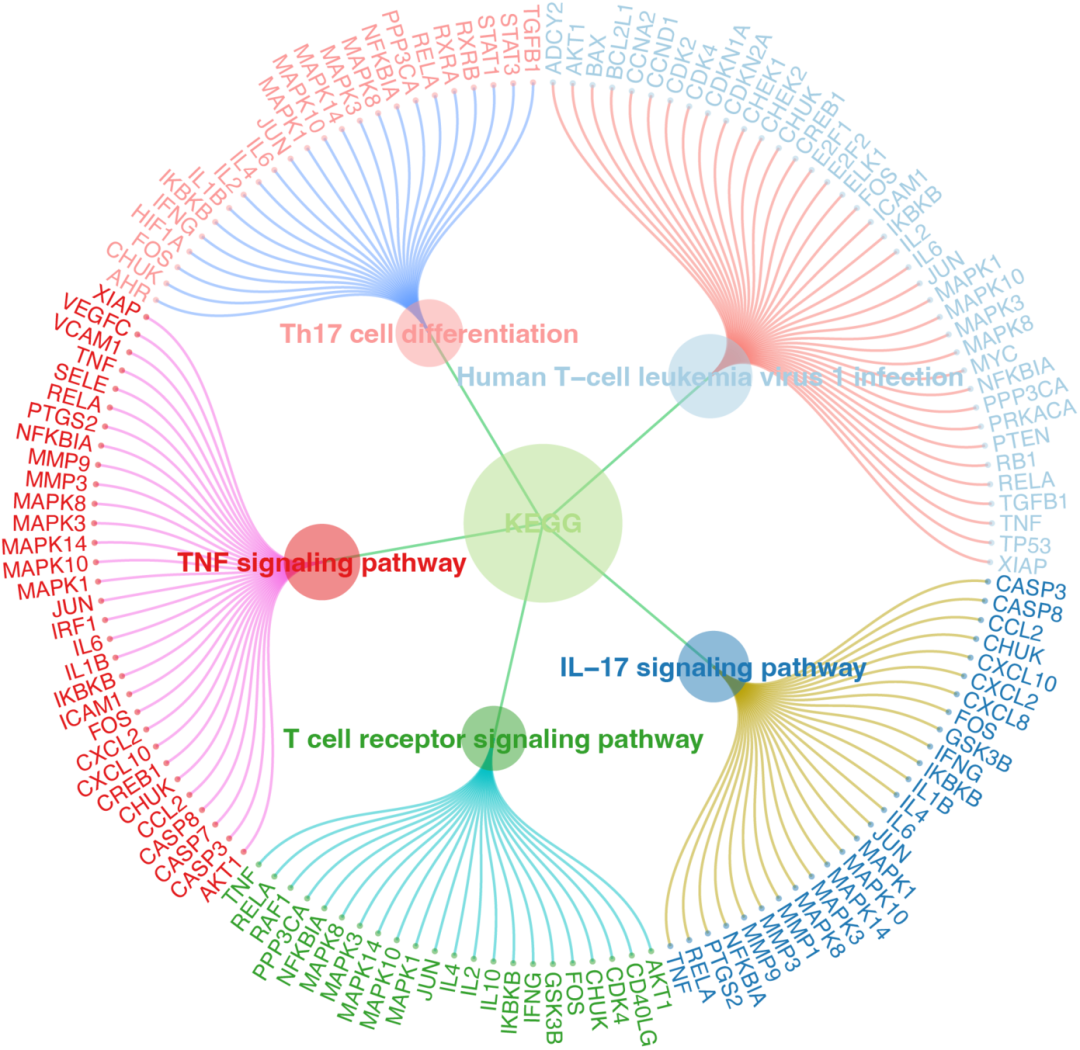
这版也不错:
bubble_plot(KK)
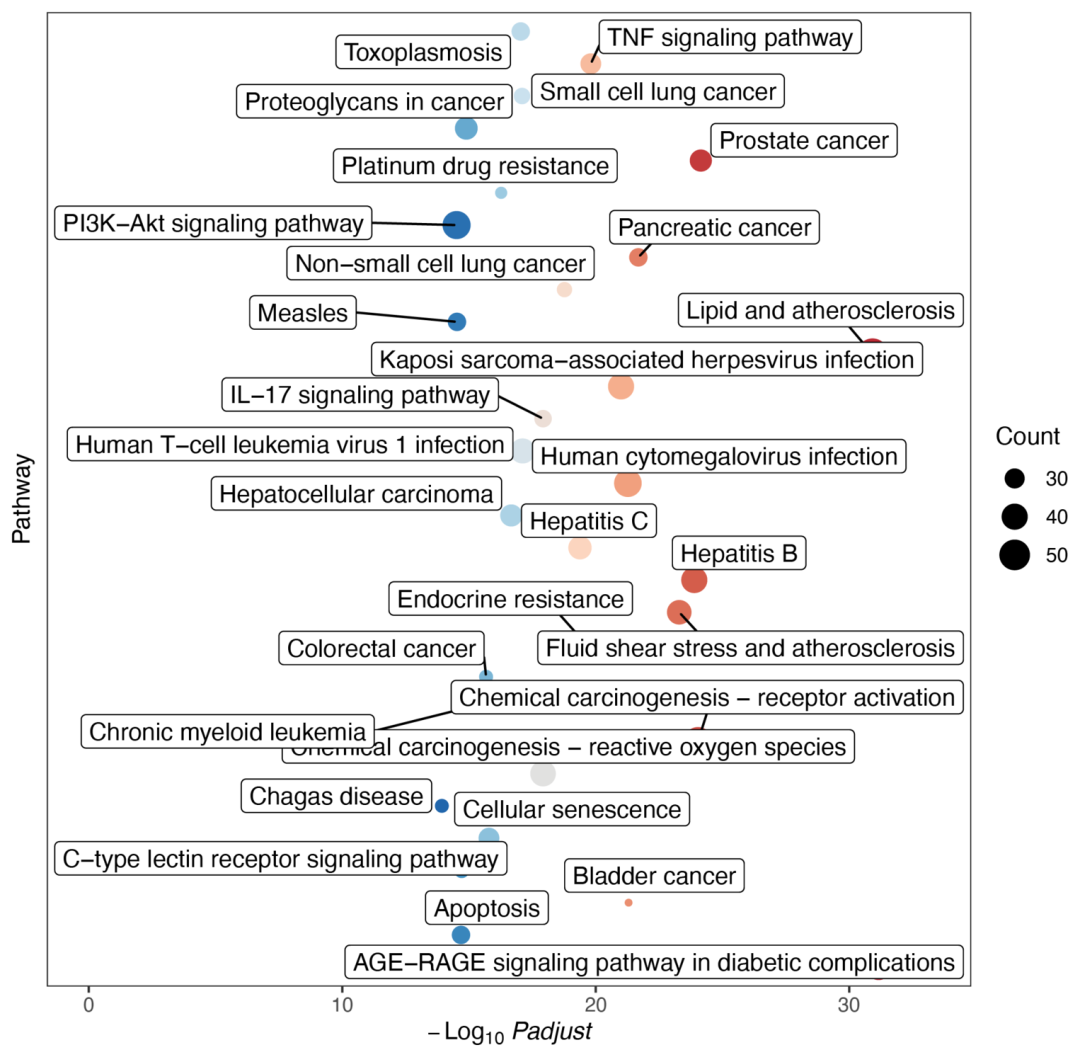
富集桑基图:
data(kegg.filter, package = "TCMNP")
#Because not all pathways and genes are what we want to display, and displaying too many genes at the same time will cause text overlap.
#The data format must be an S4 object or data frame.
#"kegg.filter" was derived from the data frame after kegg-enriched data is screened for pathways and genes.
dot_sankey(kegg.filter)
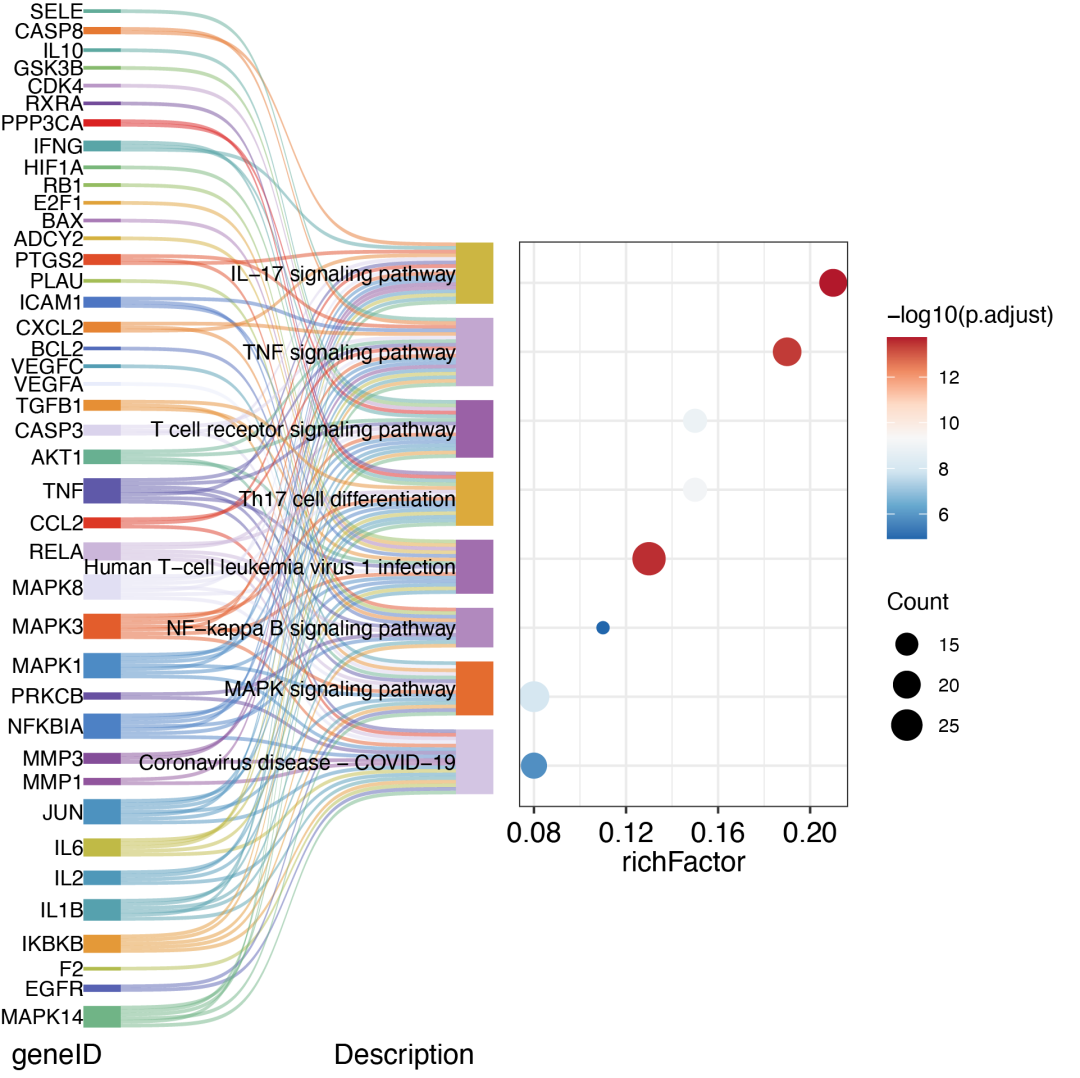
10.药物成分靶点网络桑基图:
KK2 <- KK %>% mutate(richFactor = Count / as.numeric(sub("/\\d+", "", BgRatio)))
path <- separate_rows(KK2@result, geneID, sep = "/")
data_sankey <- left_join(xfbdf, path,
by = c("target" = "geneID"),
relationship = "many-to-many"
) %>%
distinct() %>%
drop_na() %>%
sample_n(30, replace = FALSE) %>%
as.data.frame()
tcm_sankey_dot(data_sankey)
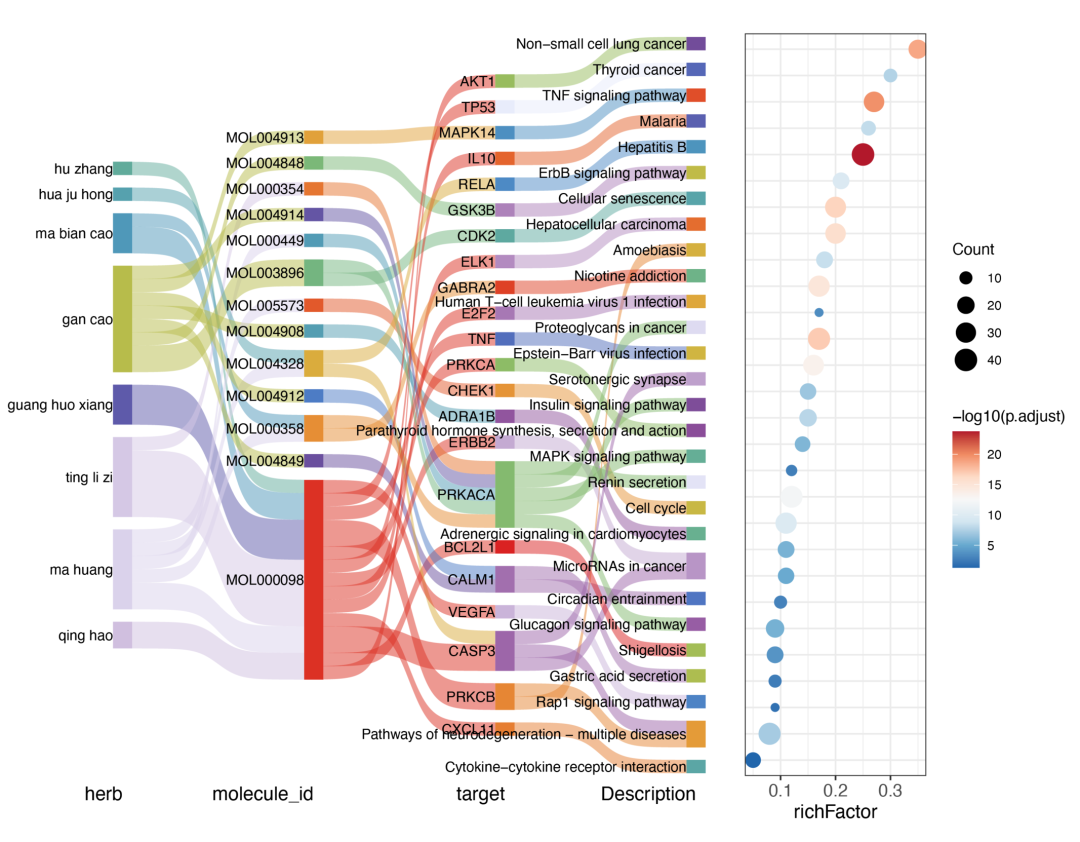
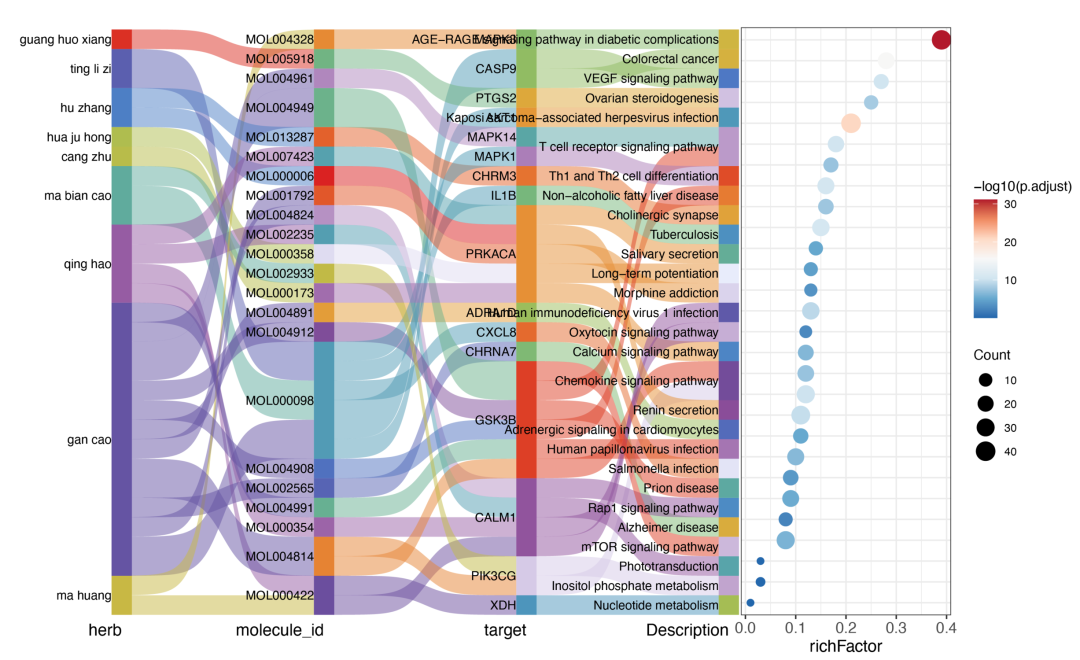
冲积图:
tcm_alluvial_dot(data_sankey)
11.蛋白互作网络ppi:
data(string, package = "TCMNP")
ppi_plot(string,
label.degree = 1,
nodes.color = "Spectral",
label.repel = TRUE)

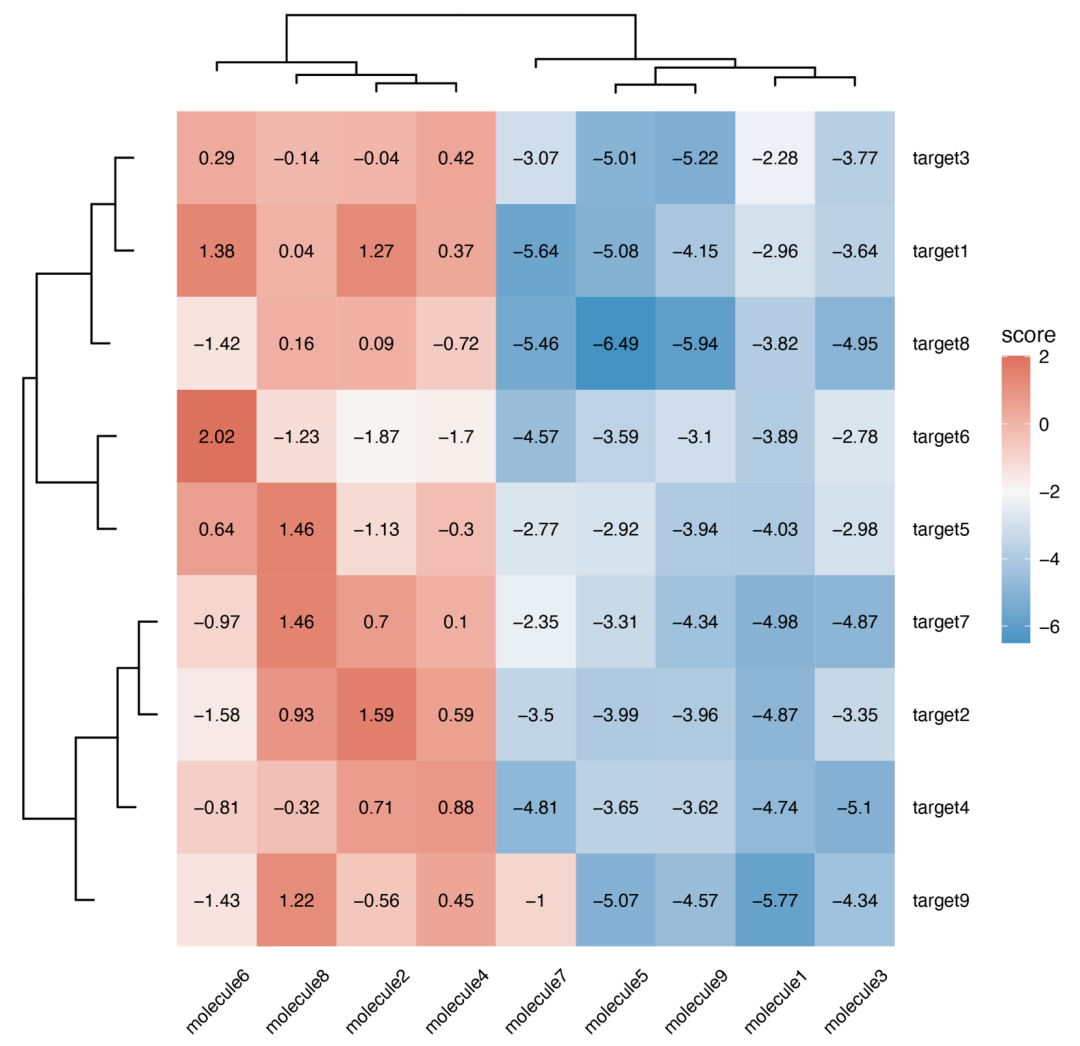
12.分子对接可视化
data <- matrix(rnorm(81), 9, 9)
data[1:9, seq(1, 9, 2)] <- data[1:9, seq(1, 9, 2)] - 4
colnames(data) <- paste("molecule", 1:9, sep = "")
rownames(data) <- paste("target", 1:9, sep = "")
data <- round(data, 2)
dock_plot(data)
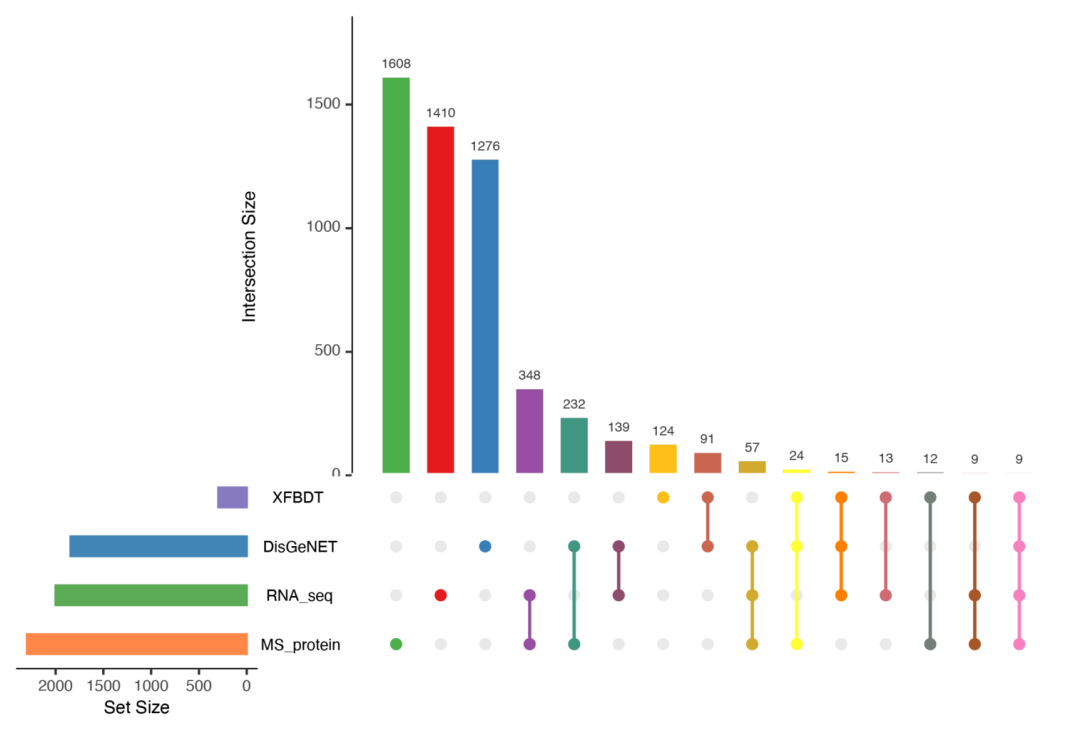
13.交集upset图:
venn_plot(venn_data, type = 2)
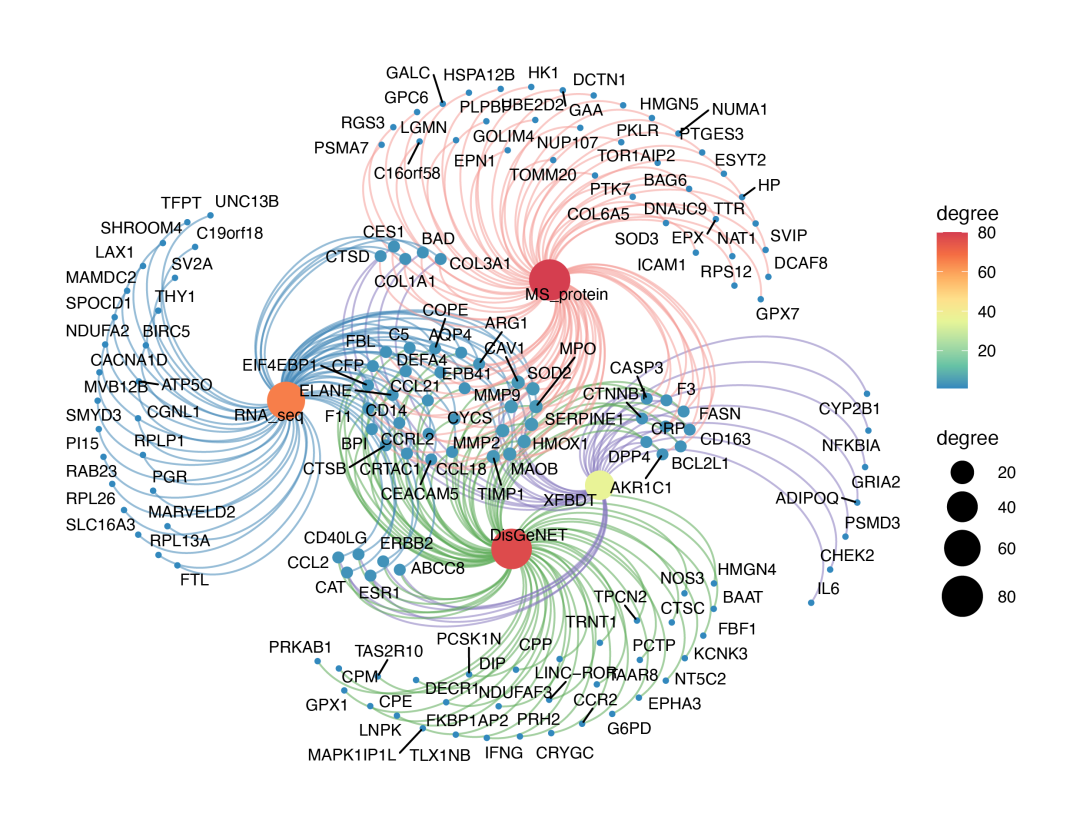
韦恩交集网络图:
data(venn_data, package = "TCMNP")
gene <- names(sort(table(venn_data$gene), decreasing = TRUE))[1:50]
data <- venn_data[venn_data$gene %in% gene, ]
data2 <- sample_n(venn_data, 100) %>% rbind(data)
venn_net(data2, label.degree = 1)
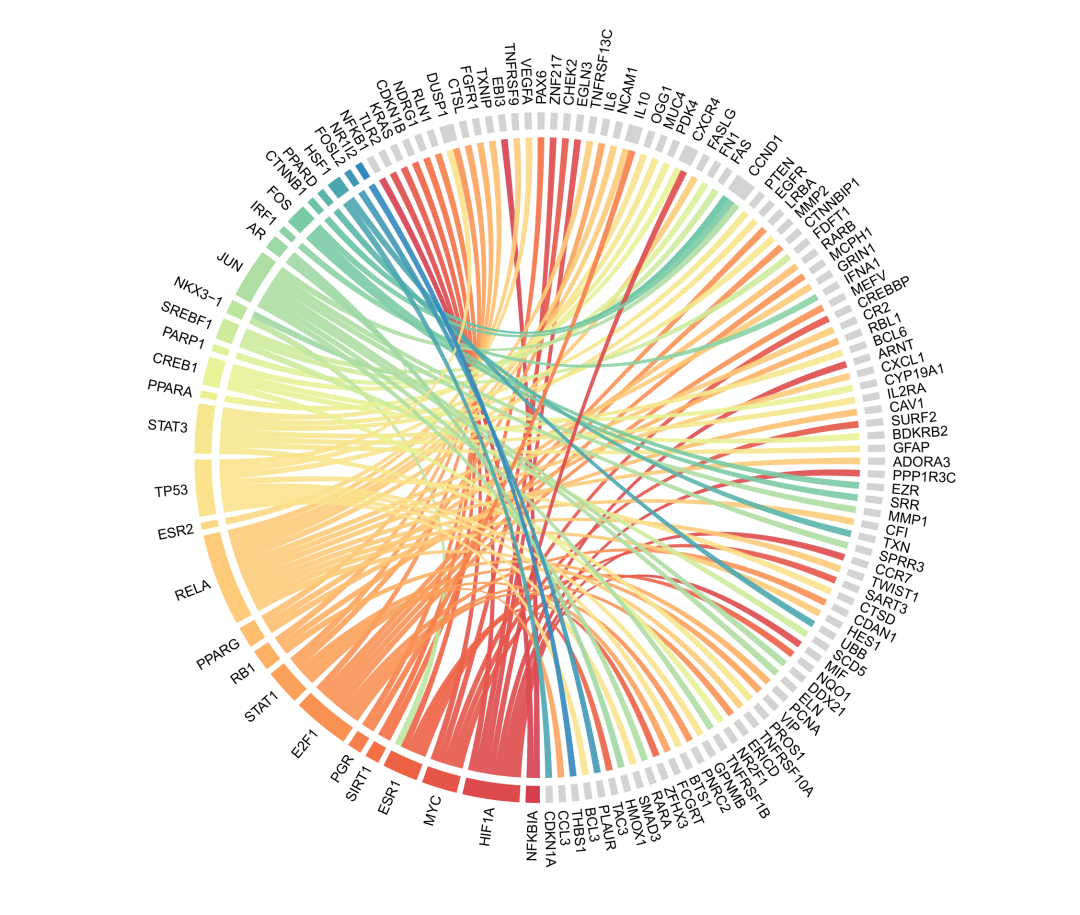
14.转录因子网络图:
# Visualization of filtered transcription factor data
data(xfbdf, package = "TCMNP")
tf_data <- tf_filter(xfbdf$target)
set.seed(1234)
data <- tf_data[, 1:2] %>%
distinct() %>%
sample_n(100)
tf_cirplot(data, color = "Spectral")
15.etcm数据库查询:
data("mahuang", package = "TCMNP")
data <- etcm(mahuang, herb = "ma huang")
head(data)
# herb molecule target QED
# ma huang Kaempferol ACTB 0.9643
# ma huang Kaempferol AHR 0.9643
# ma huang Kaempferol AKR1C1 0.8621
# ma huang Kaempferol AKT1 0.963
# ma huang Kaempferol ATP5A1 0.9643
# ma huang Kaempferol ATP5B 0.9643
功能好多
已经可以通关网药了
收藏试试
做点常规分析或者也能用

腾讯云面向开发者汇聚海量精品云计算使用和开发经验,营造开放的云计算技术生态圈。
更多推荐
 7
7 0
0- 0



 已为社区贡献13条内容
已为社区贡献13条内容

所有评论(0)