iMeta高引论文 | 刘永鑫/陈同/李国梁/陈实富/文涛-EasyMetagenome: 用户友好且灵活的宏基因组测序数据分析流程
点击蓝字 关注我们易宏基因组(EasyMetagenome):用于微生物组研究中用户友好且灵活的宏基因组测序数据分析的流程根据2026年1月公布的ESI高被引论文引用筛选阈值情况,本文远超2025年微生物学高被引入选阈值7,属于高被引论文 (Top 1%)。iMeta主页:http://www.imeta.science研究论文● 期刊: iMeta(IF 33.2, 中科院双一区Top)● 文章
点击蓝字 关注我们
易宏基因组(EasyMetagenome):用于微生物组研究中用户友好且灵活的宏基因组测序数据分析的流程

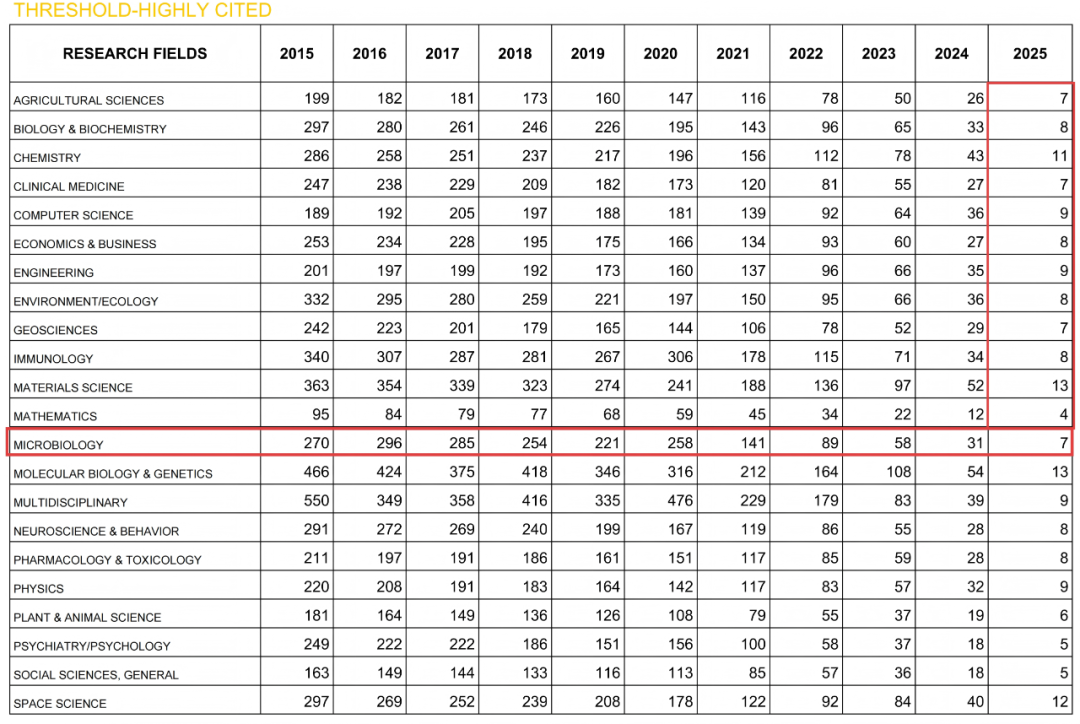
根据2026年1月公布的ESI高被引论文引用筛选阈值情况,本文远超2025年微生物学高被引入选阈值7,属于高被引论文 (Top 1%)。

iMeta主页:http://www.imeta.science
研究论文
● 期刊: iMeta(IF 33.2, 中科院双一区Top)
● 文章被引(Dimensions截至2026年1月17日):28
● 原文链接DOI: https://doi.org/10.1002/imt2.70001
● 2025年2月14日,中国农业科学院深圳农业基因组研究所刘永鑫团队联合国内多团队在iMeta在线发表了题为“EasyMetagenome: A user-friendly and flexible pipeline for shotgun metagenomic analysis in microbiome research”的文章。
● 宏基因组(EasyMetagenome)是一款用户友好的宏基因组分析流程,专为全面的微生物组数据分析设计,支持质量控制、宿主去除、基于读长、基于组装、分箱、基因组及泛基因组分析。它提供可自定义设置、数据可视化以及参数说明。未来,该流程将持续更新迭代,致力于为用户提供便捷高效的宏基因组数据分析解决方案。易宏基因组可在https://github.com/YongxinLiu/EasyMetagenome免费获取。
● 第一作者:白德凤、陈同、荀佳妮、马闯、罗豪、杨海飞
● 通讯作者:刘永鑫(liuyongxin@caas.cn)、高云云(gaoyunyun@caas.cn)、李国梁(guoliangli2016@gmail.com)、陈实富(chen@haplox.com)、文涛(taowen@njau.edu.cn)、陈同(chentong_biology@163.com)
● 合作作者:曹晨、曹晓峰、崔建洲、邓园萍、邓招超、董雯昕、董文学、杜娟、方群凯、方伟、方玥、傅芳田、傅敏、符意甜、高鹤、葛菁萍、宫庆龙、辜伦达、郭鹏、郭宇豪、海棠、刘昊、何杰强、贺子洋、侯辉宇、黄灿、季帅、姜昌海、姜桂来、江玲娟、金灵、阚玉贺、康达、寇谨、林嘉龙、李长超、李翀、李馥伊、李立威、李妙、李心、黎烨、李政弢、梁晶、林永新、刘长振、刘丹妮、刘凤琴、刘佳、刘天睿、刘婷婷、刘新元、刘亚群、刘帮艳、刘明皓、娄文博、栾亚宁、罗圆圆、吕虎杰、马腾飞、麦宗炯、莫家远、牛东泽、潘卓、亓合媛、石展耀、宋春娇、孙馥香、孙岩、田思惠、万锈琳、王国良、王红阳、王红玉、王焕焕、王婧、王君、王康、王乐莅、王绍坤、汪欣龙、王瑶、肖祖飞、邢卉春、许艺凡、阎姝彦、杨丽、杨松、杨元明、姚小芳、Salsabeel Yousuf、于浩、雷宇、袁峥嵘、曾美尹、章春芳、张春鸽、张慧敏、张敬、张娜、张天缘、张毅波、张玉鹏、张政、周明达、周远平、朱成帅、朱琳、朱跃、朱志豪、邹洪琴、左安娜、董文轩
● 主要单位:中国农业科学院深圳农业基因组研究所、中国中医科学院、安徽农业大学、青岛农业大学、南京医科大学、清华大学、新加坡国立大学、湖南农业大学、浙江大学海洋学院、农业农村部环境保护科研监测所、西藏民族大学医学院西藏自治区高原病分子遗传机制与干预研究重点实验室、瑞典卡罗琳斯卡医学院、浙江工业大学、浙江农林大学、北京林业大学、杭州微致生物科技有限公司、中南大学、广东省科学院微生物研究所、黑龙江大学、吉林农业大学、圣湘生物科技股份有限公司、河北科技大学、山西大同大学、西交利物浦大学、西北农林科技大学园艺学院、墨尔本大学、东京大学、赫尔辛基大学、上海海事大学、苏州大学、北京协和医院、香港理工大学、潍坊学院、北京工业大学、兰州交通大学、香港中文大学生命科学院、阿尔伯塔大学、东北师范大学、广西医科大学第二附属医院、虹摹生物科技(上海)有限公司、密歇根大学公共卫生学院、中国科学院南京土壤研究所、浙江大学、广西大学、福建师范大学、河北工程大学、海普洛斯、河南农业大学、南开大学、中国中医科学院中医药健康产业研究所、首都医科大学附属北京地坛医院传染病研究所、韩山师范学校、中国科学院微生物研究所、伦敦帝国理工学院、兰州大学、中山大学附属第五医院、常州大学、河南省肿瘤医院、北京师范大学、德阳市人民医院、新方向生物科技(天津)有限公司、中国科学院植物研究所、北京市农林科学院生物技术研究所、中国中医科学院中药资源中心、安徽科技学院、中国农业科学院植物保护研究所、中国环境科学研究院、中国疾病预防控制中心、扬州大学、中国科学院亚热带农业生态研究所、中国林业科学研究院生态保护与修复研究所、中国科学院城市环境研究所、首都医科大学附属北京地坛医院肝病三科、广州中医药大学、西北农林科技大学、北京林业大学生物科学与技术学院、石河子大学、北京联合大学、中国农业科学院茶叶研究所、同济大学、广东医科大学、哈尔滨工业大学、广东工业大学、中国农业科学院农业资源与农业区划研究所、南方医科大学、普渡大学、南京农业大学、江西农业大学等
亮 点
● 易宏基因组(EasyMetagenome) 提供多种安装选项,并附有详细的解释答疑;
● 易宏基因组包含详细的参数说明,可适应多种数据场景,确保其灵活性与适应性;
● 易宏基因组支持从原始数据到下游的全面分析,包括基于读长、基于组装、分箱及基因组分析,同时能够生成可用于发表的可视化结果;
●易宏基因组作为开源项目开发,鼓励社区合作。可通过 https://github.com/YongxinLiu/EasyMetagenome 获取和参与贡献。
摘 要
在宏基因组学研究领域,鸟枪法测序已成为一项核心技术,使研究人员能够以高分辨率从分类和功能层面深入解析微生物群落。这一方法为揭示微生物的多样性、相互作用及其在健康与疾病中的作用提供了重要见解。然而,数据处理的复杂性和研究结果的可重复性仍是学术界面临的主要挑战。为此,我们开发了易宏基因组(EasyMetagenome),一个用户友好的分析流程,支持多种分析方法,包括质量控制与宿主序列去除、基于读长、组装和分箱的方法,以及高级基因组分析功能。该流程提供了用户可自定义的设置、丰富的数据可视化功能,以及详细的参数说明,确保其能够灵活应对不同场景数据处理的需求。展望未来,我们将致力于解决宿主污染问题,优化适配三代测序数据的工作流程,并整合深度学习和网络分析等前沿技术,以进一步提升对微生物组研究的洞察力和数据准确性。易宏基因组可在https://github.com/YongxinLiu/EasyMetagenome免费获取。
视频解读
Bilibili:https://www.bilibili.com/video/BV1DWAkeMERd/
Youtube:https://youtu.be/sdxzL9thnDg
中文翻译、PPT、中/英文视频解读等扩展资料下载
请访问期刊官网:http://www.imeta.science/
全文解读
引 言
微生物,包括细菌、古菌、真菌、病毒和原生生物,广泛存在于自然环境中。“微生物组”一词通常指特定宿主或环境中的微生物群落及其基因组。微生物组研究显著推动了多个领域的发展,包括农业、食品科学、生物技术、生物经济学、动植物营养与健康,尤其在人类医学领域取得了重要突破。自从首次发布来自环境微生物群样本的鸟枪法宏基因组数据以来,微生物组研究在数据生成和方法学进步方面呈现爆炸式增长。
与通常针对特定基因区域(如16S rRNA基因)的扩增子测序不同,鸟枪法宏基因组学测序(也称短读长测序、第二代测序、下一代测序或高通量测序)能够捕获样本中的全部DNA。它能够提供微生物群落完整分类组成和功能特征的相对无偏见视图。此外,该技术还能重建宏基因组组装基因组(metagenomic assembled genomes, MAGs),以高分类学分辨率揭示微生物多样性,能精确到物种水平,甚至在某些情况下达到菌株水平。因此,鸟枪法宏基因组测序已成为微生物组研究中的变革性工具,极大地加深了我们对人类及其他环境中微生物群落的理解。尽管第三代长读长测序技术的发展在一定程度上弥补了短读长测序的局限性,但其较高的错误率和较低的通量使得大多数研究仍依赖于混合策略(结合长读长和短读长数据)进行错误校正。因此,第二代宏基因组测序仍在微生物组研究中占据关键地位。
在从目标微生物群落生成鸟枪法宏基因组数据后,通常的分析流程从质量控制和宿主DNA去除开始,随后对微生物组的分类学、功能和基因组特征进行解析,以深入了解其生物学基础。针对分析流程的各个阶段,已经开发了多种工具、R包和流程。例如,fastp 或 Trimmomatic用于基本的质量控制,Bowtie2或KneadData (https://huttenhower.sph.harvard.edu/kneaddata) 用于移除映射到宿主参考数据库的序列,Kraken2和 Bracken用于分类学分析,HUMAnN3用于生成基因家族及 MetaCyc代谢通路,MEGAHIT或 MetaSPAdes用作宏基因组组装工具,而 MetaWRAP可从宏基因组中提取宏基因组组装基因组。此外,还有大量的R包可用于探索微生物群落的多样性、结构及其潜在功能。然而,这些工具或R包均包含多种参数或参考数据库,这些因素可能影响最终结果及后续分析,给不同研究间的可比性和可重复性带来了挑战。因此,制定标准化的数据处理流程显得尤为重要,以确保数据的一致性、结果的可靠性,并促进不同研究间发现的可重复性。
近年来,尽管已有多个工作流程被开发用于微生物组研究,但大多数工作流程都是为特定分析任务设计的,缺乏适用于更广泛数据探索的整体性方法。此外,这些工作流程通常缺乏详尽的报告、灵活的参数调整选项或有意义的下游可视化功能,而这些对于结果的解读和研究的可重复性至关重要。例如,Sunbeam 主要专注于基础分析,包括数据预处理、基于读长的分析以及基于组装的分析;MetaSAMS 工作流程则主要用于分类学分析和功能分析(基于读长的分析);而 MEGAnnotator2、SqueezeMeta和 ATLAS工作流程分别被开发用于从宏基因组序列中重建MAGs(基于组装和分箱的分析)。METABOLIC 则专注于MAGs的功能特性分析,包含与MAGs分析相关的工作流程。此外,一些在线平台,如 MicrobiomeAnalyst 2.0和 Wekemo Bioincloud,提供了用于统计分析和鸟枪法数据矩阵分析的实用工具。然而,这些平台在处理大型数据集时存在局限性,尤其在鸟枪法宏基因组学中,数据量通常从数百GB到数TB不等,这使得在线处理变得尤为困难。
在这里,我们介绍了 EasyMetagenome,一个易于部署和配置的流程,支持基于读长、基于组装和分箱的分析,以及深入的原核基因组分析,生成适合发表的可视化结果。EasyMetagenome 包括四个主要组成部分:软件和数据库安装、数据分析流程、统计与可视化、以及专门的问答部分。几乎所有步骤都是可配置的,都具有合理的默认设置,允许在无需大量参数调节的情况下快速部署。此外,还提供了详细的软件参数说明,使其能够适应各种数据分析场景。
结 果
易宏基因组(EasyMetagenome)流程概述
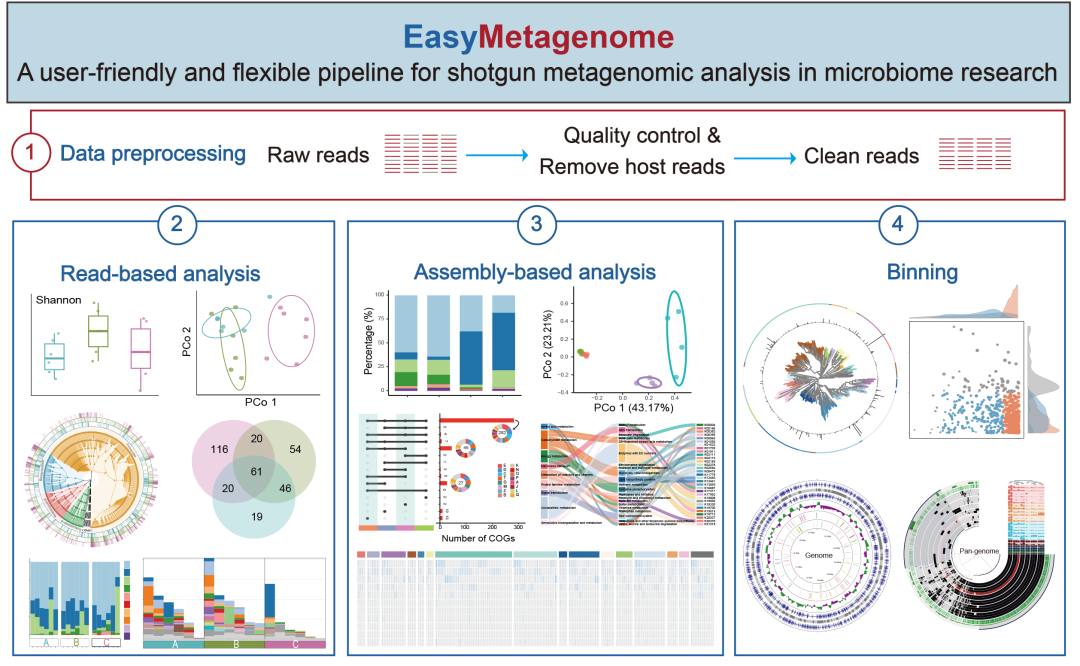
易宏基因组包含四个主要组成部分:软件和数据库安装、数据分析流程、统计与可视化,以及专门的问答部分。每个组成部分都是独立的 shell 脚本,并内置有问答功能,帮助用户理解和修改流程(图1)。目前,它提供了便捷的工作流程,用于数据预处理、基于读长的分析、基于组装的分析和分箱,采用了本领域一系列先进的软件和数据资源。
在根据我们的安装说明完成必要的软件和数据库安装后,用户可以输入双端宏基因组测序文件以启动分析。易宏基因组是完全集成的端到端流程,涵盖从原始读长处理到最终数据表和适合发表的图表(图1)。首先,易宏基因组会对原始序列/读长进行质量控制和宿主基因组去污染处理。然后,清洁后的读长可以通过基于读长的分析进行分类和功能注释,或者通过基于组装的分析进行组装、基因预测、功能基因注释和分类学分类。此外,组装结果会通过 metaWARP 进行分箱和优化,随后进行去重和恢复的MAGs评估。在统计与可视化部分,我们还提供了多种R语言配置文件,便于用户生成和自定义图表。截至2024年11月27日,易宏基因组在Github上的星标数为351,Fork数为225(https://github.com/YongxinLiu/EasyMetagenome)。
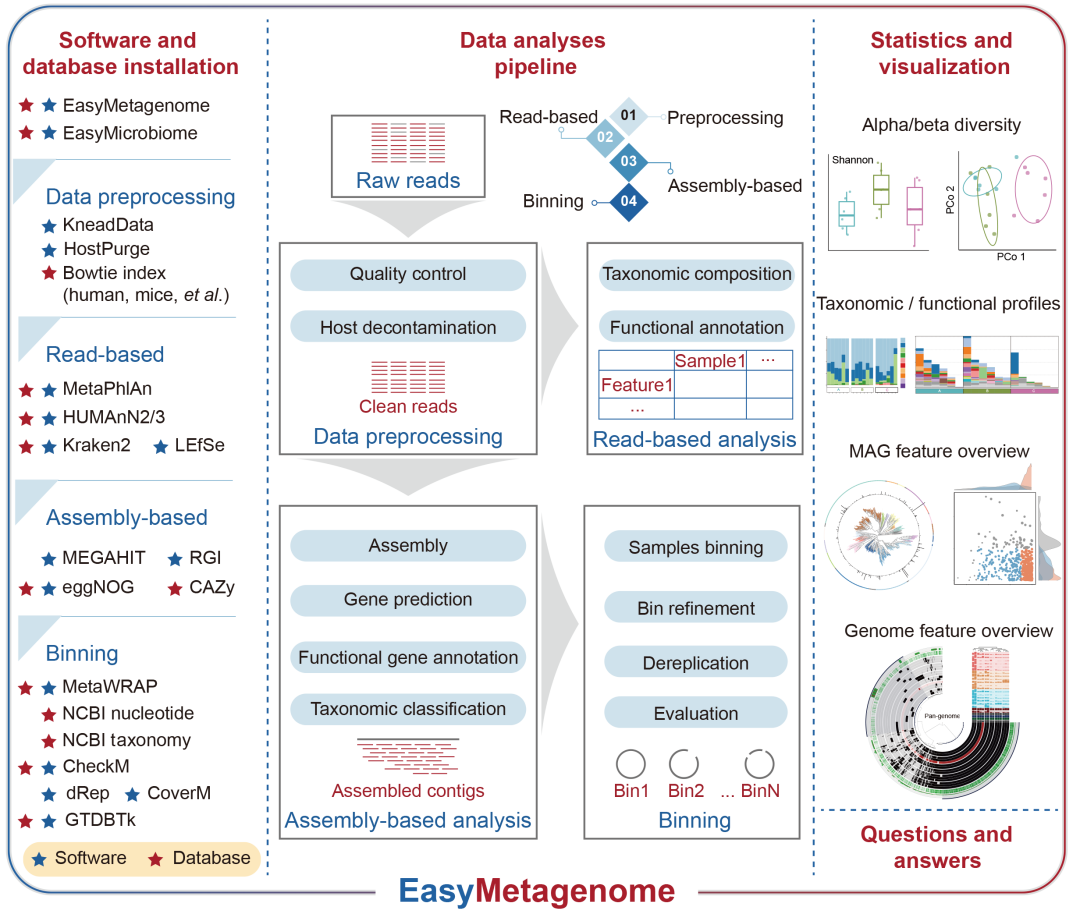
图1. EasyMetagenome工作流程
该工作流程包含四个主要组成部分,包括软件和数据库安装、数据分析流程、统计与可视化以及问答部分。
软件安装和数据库部署
EasyMetagenome 的安装创建的生物信息学分析环境,包含超过150种常用的生物信息学软件和数据库,具体包括41个软件、23个数据库和90多个核心R包(图2)。为了简化安装过程,我们为每个软件提供了至少两种安装方法,减少了研究人员的工作量,提高安装成功率,以便他们集中精力处理更紧急的任务。该流程目前包括四种数据预处理工具、10种基于读长的分析工具、15种基于组装的分析工具和12种分箱工具(图2A,表1)。
对于常用的公共数据库,我们提供了至少两个可访问的备份链接(如FTP服务器和百度网盘)。此外,我们还提供了详细的数据库创建定制说明。例如,我们不仅提供了预构建的鼠类(C57BL/6NJ)和人类(CRCh37)索引数据库,还提供了使用 Bowtie2 构建自定义索引数据库的详细步骤指南。为了帮助用户根据服务器内存和使用需求选择合适的数据库,我们提供了三种常用的 Kraken2 数据库版本:16GB版本的标准数据库(包含原生生物和真菌)(PlusPF16G),69GB版本的标准数据库(包含原生生物和真菌)(PlusPF),以及144GB版本的标准数据库(包含原生生物、真菌和植物)(PlusPFP)。我们还提供了构建自定义微生物数据库的指导(图2B)。
随后,我们提供了一系列R语言脚本,用于生成各个领域的可发布图表(图2C)。对于差异分析,我们使用了21个R包来检测显著差异并可视化结果。多样性分析支持23个R包,涵盖了α多样性、β多样性、稀释曲线和组成分析。群落比较采用了8个关键包进行排序、聚类和多样性度量。生物标志物识别涉及22个包,利用了机器学习和统计建模方法。网络分析依赖于11个包来识别和可视化数据集中的关系。此外,18个包辅助数据处理、可视化和注释,确保提供了全面的分析方法。
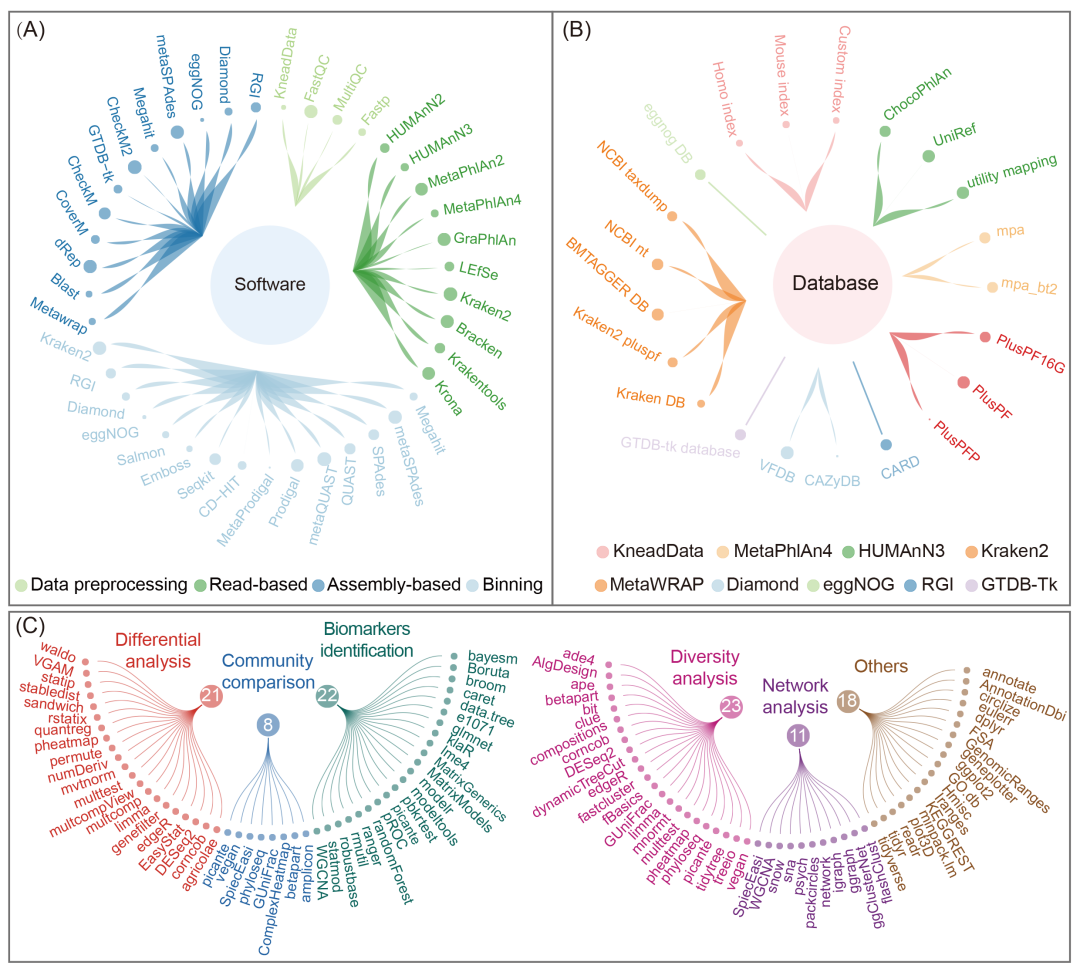
图2. EasyMetagenome 中包含的软件、数据库和 R 包
(A) EasyMetagenome 流程中使用的软件,涵盖数据预处理、基于读长的分析、基于组装的分析和分箱分析。(B) 与每个软件相关的标准或自定义数据库。(C) 用于差异分析、群落比较、生物标志物识别、多样性分析、网络分析等的 R 包。
数据分析流程设计
为了指导分析过程,我们在流程中提供了清晰的工作目录和文件结构(图3)。第一步是对原始序列数据进行预处理,这是去除低质量读长和宿主相关序列污染的关键步骤,确保下游分析的数据更加清洁和可靠。第二步涉及使用基于读长的方法进行分类学和代谢潜力分析,这对于识别微生物的组成和功能至关重要,并为研究的生态系统提供了有价值的见解。在这一步中,我们使用Kraken2 用于微生物组成分析,使用HUMAnN2 或 HUMAnN3 用于功能分析,该方法简单且应用广泛,但可能漏掉当前数据库中未注释的未知基因。
组装步骤是整个流程中的关键部分,其中高质量的读长通过 MEGAHIT 或 metaSPAdes 等工具组装成重叠群。MEGAHIT 用于优化快速组装大型、复杂的宏基因组数据集,并且所需计算资源较少,而 metaSPAdes 则能生成较长且达到菌株水平的重叠群(contigs),但需要更多的计算资源。一旦重叠群被组装完成,接下来可以进行组装评估、基因预测、冗余基因去除以及基因丰度定量。宏基因组数据集通常包含数百万个基因,这些基因随后可以合并为功能类别,如 KEGG 同源基因(KEGG Orthology, KO)、同源基因簇(Clusters of Orthologous Genes, COG)、碳水化合物活性酶(Carbohydrate Active Enzyme, CAZy)、抗生素抗性基因(Antibiotic Resistance Genes, ARG)等。
在宏基因组分箱步骤中,重叠群被聚类成基因组箱。这可以单独对每一组组装的重叠群进行处理,或通过将来自多个样本的重叠群汇集在一起,并将每个样本映射回共有的重叠群目录来完成。像 metaWRAP 这样的工具可以促进这一过程,通过重叠群分箱生成 MAGs。在宏基因组学研究中,分箱使得已知和未知基因组的重建成为可能,为微生物群落提供了全面的描述,并为进一步的分析奠定了基础。
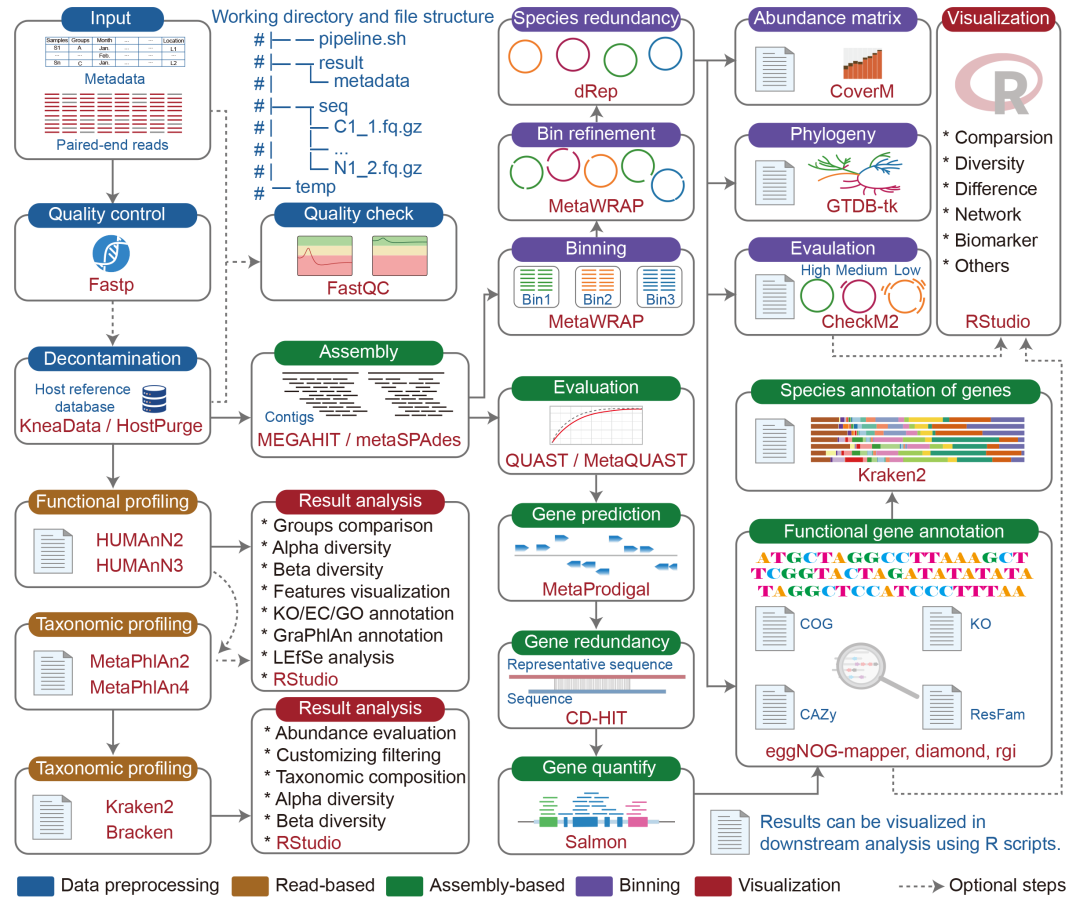
图3. EasyMetagenome 分析流程概述
EasyMetagenome 流程包括基于读长、基于组装和基于分箱分析的数据预处理,并提供可用于数据可视化的文件。虚线表示可选步骤,软件以红色突出显示。
基于读长分析的分类和功能组成
在这里,我们展示了基于读长分析的结果,利用来自百岁老人肠道微生物组研究的数据,展示了分类学和功能组成的多样性。分类学组成使用 Kraken2(图S1)和 MetaPhlAn4(图4)进行评估,随后进行 α 多样性和 β 多样性分析,并使用 HUMAnN3(图4)评估功能组成。我们提供了多种可视化风格,以帮助阐明和丰富这些结果的解释,提供了关于群落结构和功能动态的详细、多维度视角(图4)。
对于宏基因组数据的分类学注释,Kraken2 和 MetaPhlAn4 提供了各具特色且互补的方法。Kraken2 使用基于 k-mer 的方法进行分类,兼容如 PlusPF16G、PlusPF 和 PlusPFP 等数据库(图2B)。为了优化分类丰度估计,Bracken 在 Kraken2 的结果基础上,通过概率性地重新分配读长来改进结果。另一方面,MetaPhlAn4 通过使用特定类群的标记基因目录(如 mpa、mpa_bt2)来实现分类学分析,提供了一个聚焦于基因的分类学注释方法。对于功能潜力分析,HUMAnN3 通过使用 UniRef、ChocoPhlAn 以及一组实用的映射文件来对功能基因和通路进行映射。
为了可视化物种和功能组成,我们在百岁老人、老年人和年轻人群体中按门水平分析细菌分布,突出每个样本中物种的存在和相对丰度(图4A)。韦恩图展示了各组之间的共有和特有分类单元(图4B),而堆叠条形图则比较了组间和组内的分类分布(图4C)。我们计算了 α 多样性指标,包括 Chao1、ACE、Shannon、Simpson、Inverse Simpson,也计算了多个β 多样性指标(Bray-Curtis、Euclidean、Jaccard、Manhattan),并对各样本和组进行了分析(图4D、E)。为了分析组间的物种差异,我们使用 STAMP 图进行比较,通过条形图展示了分类单元或功能通路的丰度(图4F)。微生物物种对这些功能通路的分层贡献进一步阐明了群落组成和功能(图4G)。图S1A、B 中也进一步展示了Kraken2的结果。
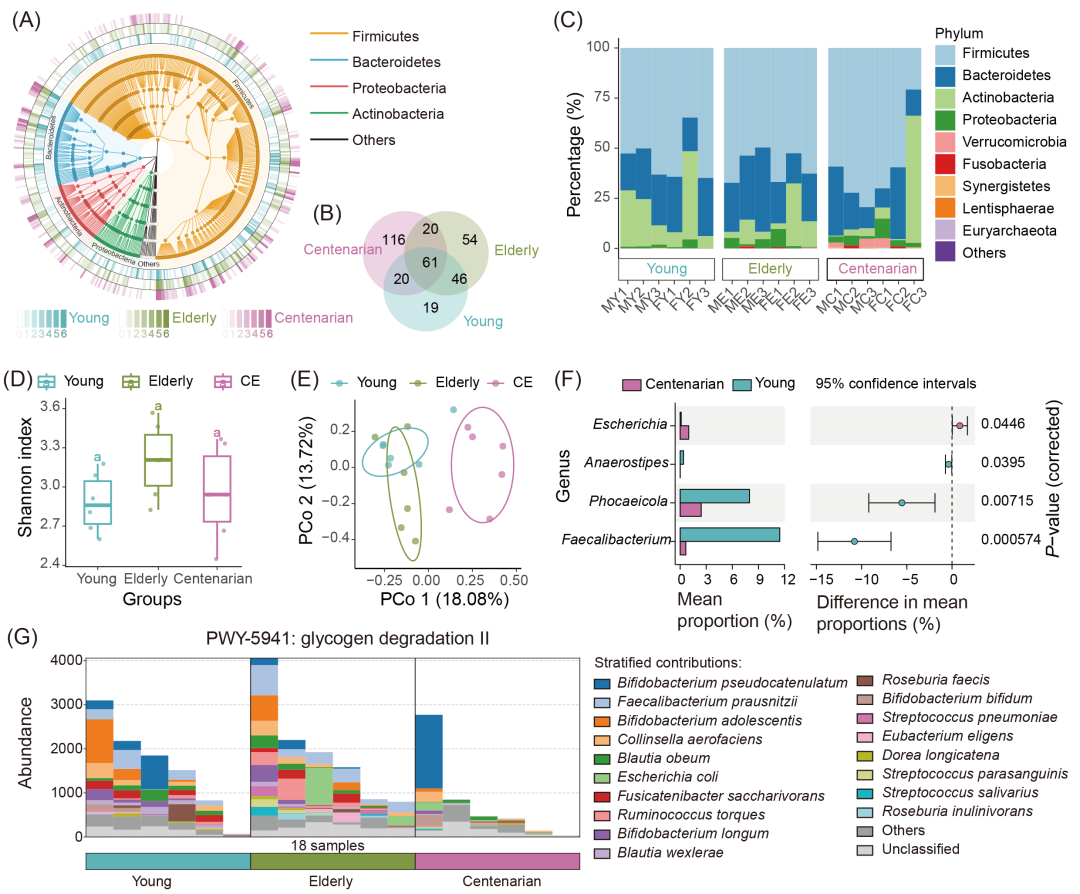
图4. 使用 EasyMetagenome 对人类肠道微生物组样本进行基于读长的分析得到的分类和功能组成
(A)各组微生物分类组成概览。内圈线条颜色表示门。外圈条形阴影表示每组六个代表性样本中分类群的存在或缺失。(B) 韦恩图显示三个组之间独特和共有的分类群。(C) 各组和样本在门级别的分类组成。(D) 各组的香农多样性指数,字母表示显著的组间差异(P ≤ 0.05,ANOVA,Tukey’s HSD)。(E) 基于 Bray-Curtis 相异性(P < 0.001,PERMANOVA 和 ADONIS 检验)的主坐标分析(PCoA)。(F) STAMP 图显示年轻组和百岁组之间的属级差异。(G) 各组和样本中功能通路 PWY-5941 的丰度,包括特定微生物物种的分层贡献。(A-F) 代表使用 MetaPhlAn4 进行的物种注释分析结果。(G) 代表通过 HUMAnN3 进行的功能潜力分析。原始数据来自于NCBI发布 的测序项目(PRJNA675598),序列号见表 S1。
来自组装重叠群的基因功能注释及MAGs的比较分析
我们基于组装的分析能够使用一系列全面的数据库对组装重叠群中的基因序列进行功能注释,包括基因本体(Gene Ontologies, GOs)数据库、酶分类(Enzyme Commission categories, ECs)数据库、同源基因簇(Clusters of Orthologous Genes, COGs)数据库、碳水化合物活性酶(carbohydrate-active enzymes, CAZy)数据库和KEGG功能通路数据库(图5A,图S2A)。该流程提供了几个关键功能,首先,它展示了不同分类中蛋白质功能注释的分布,并可视化了它们的重叠情况(图5A)。其次,它还允许比较不同组之间功能模块的丰度和多样性,包括 COGs、KOs 和 CAZy,以探索微生物组的功能变化(图S2A)。此外,注释的功能基因按层级划分(如 KEGG),展示了这些功能在各个层级的结构化分布(图5B)。
在分箱流程之后,我们可以生成 MAGs 的质量评估、物种注释和功能概况。然后, MAGs 可以进行平均覆盖度、系统发育推断以及在不同分类学层级上的物种分布分析,展示所有样本的平均覆盖度(图S2B)或特定组中的平均覆盖度(图5C)。MAGs 的门信息通过 GTDB-tk 进行汇总(图5D,图S2C),MAGs 的完整性和污染率在 CheckM 中进行评估(图S3)。具有完整性 ≥ 90% 和污染度 ≤ 5% 的高质量 MAGs 可以用于进一步分析,以评估其分类组成、功能概况以及 CAZy(图5E)或抗生素抗性基因(图S2D)的情况。
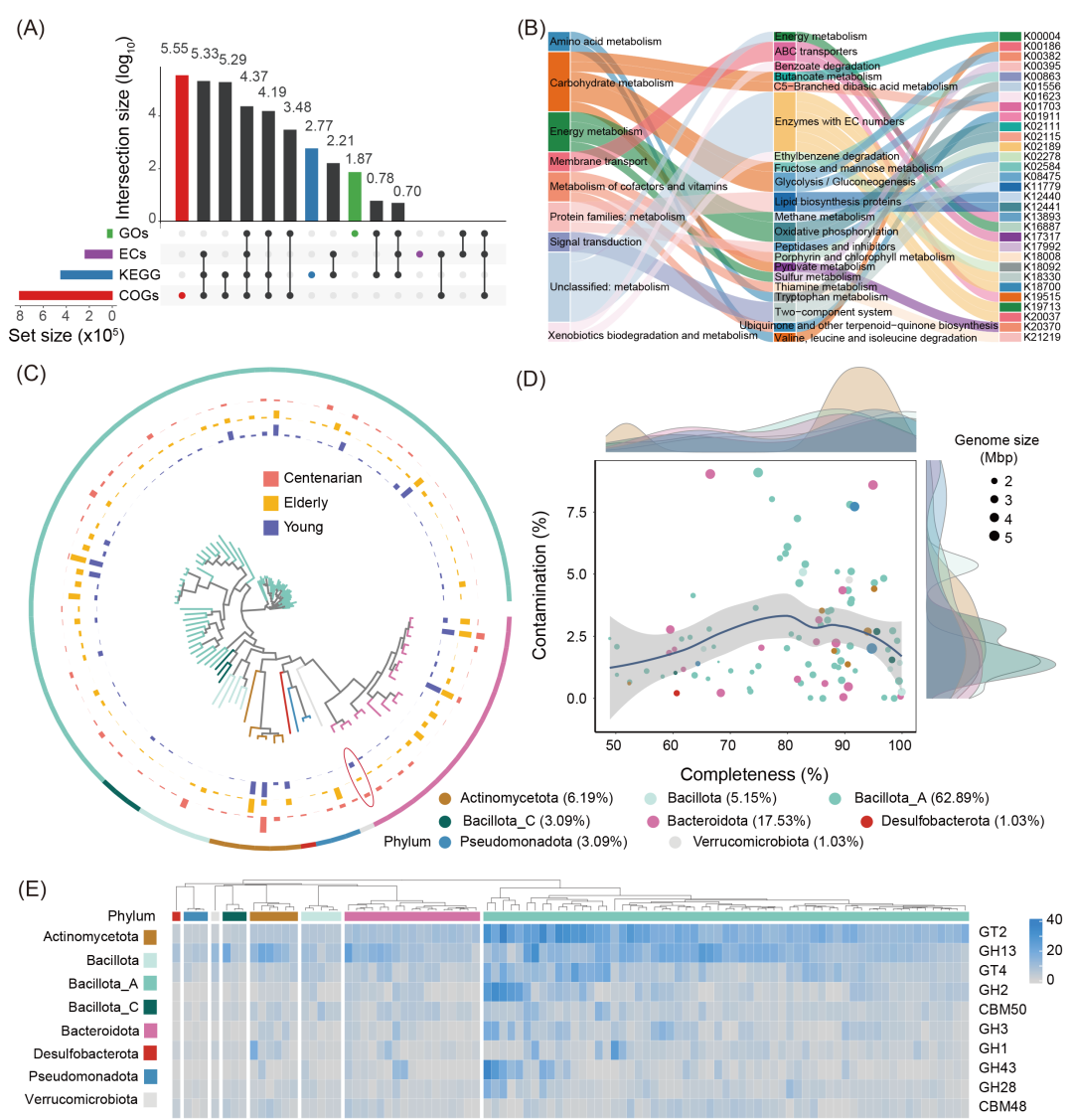
图5. 组装重叠群的功能注释以及人类肠道微生物组样本中 MAGs 的比较分析
(A) 组装的重叠群功能注释,按四个类别展示每个类别的注释蛋白数量及其重叠情况。柱状图表示各类别之间的特有(彩色)或共有(黑色)蛋白质;水平条形图展示每个类别的总功能注释蛋白数量。(B) 功能注释基因的 KEGG 分类,采用流程图可视化不同层级(初级、次级、三级)上的功能分布。(C) 不同组之间的宏基因组组装基因组(MAGs)系统发育分析与分布,突出显示代表性 MAGs 的进化关系。树状图按门进行着色,条形图展示通过 CoverM 计算的不同组中 MAGs 的平均相对丰度。(D) MAGs 的完整性与污染率分布,点的颜色表示不同门。(E) MAGs 中 CAZy 功能基因的注释,展示了 MAGs 中关键碳水化合物活性基因的分布情况。
Alistipes putredinis的特征描述及MAGs的泛基因组分析
我们的流程提供了对分离株或 MAGs 的详细功能注释和分析。在本研究中,我们聚焦于 Alistipes putredinis,这是人类肠道微生物群中的常见物种,旨在深入了解该物种的功能特征。Alistipes putredinis 的 MAG 主要在百岁老人组中被识别,显示其完整性为 96.23%,污染率为 0.67%,基因组大小为 2.08 Mb,GC 含量为 55%(图6A)。
为了检查 Alistipes putredinis MAG 与 14 个相关 Alistipes putredinis 基因组之间共有和特有的功能基因组成,我们进行了泛基因组分析(图6B)。该分析识别出了 Alistipes putredinis 泛基因组中的 3,958 个基因,涵盖了 14 个公开可用的 Alistipes putredinis 基因组和我们从 18 个宏基因组样本中注释的 Alistipes putredinis MAG。基于基因簇频率,这 15 个基因组被划分为 6 个类群,发现了 1,322 个核心基因(占 33.4%)和 1,028 个单态(singleton)基因(占 26.0%),这些单态基因是各个基因组的特有基因。根据基因簇频率(包括数量和相似度)对这 15 个基因组进行聚类,进一步将它们分为两个组和 6 个类群。每个基因组包含 1,821 到 2,323 个基因(右上角的条形图),其中核心基因占每个基因组总基因数的 57% 到 73%(图6B)。
随后,我们计算了 15 个 Alistipes putredinis 基因组之间的平均核苷酸相似性(ANI),发现 Alistipes putredinis MAG 与其他已发布基因组的相似性为 97.22% 到 99.11%(图6C)。由于基因组之间共有的基因组成能够有效预测系统发育关系,我们使用基因分布推断这些基因组之间的关系。与使用 40 个核心基因生成的聚类树状图相比,类群的聚类显示出显著差异(图6D),这表明,结合整个基因组内容而不仅仅是核心基因,可能有助于提高确定不同基因组之间关系的准确性。我们的结果还可能表明,使用整个基因组内容而不是部分核心基因,对于推断不同基因组之间的关系可能更为有利。最后,我们还通过 COG 数据库对 Alistipes putredinis 基因组进行了功能注释。我们选择了 24 个具有高 AG 比率的 COG 功能,以展示泛基因组中的功能多样性。这使我们能够观察到不同类群在每个选定 COG 功能中 CG / AG 比率的变化,为类群之间的功能差异提供了新的见解(图6E)。

图6. 在人类肠道微生物组示例中的宏基因组组装基因组挖掘
(A)Alistipes putredinis 基因组图(ncRNA、COG 注释、GC 含量和 GC 偏度)。从内到外,第一个圆圈显示基因组大小(1 基因组大小);第二个圆圈显示正链基因的 COG 功能,每种颜色代表一个功能分类(2 正链功能);第三个圆圈显示反链基因的功能(3 反链功能);第四个圆圈显示 GC 偏度((G - C) / (G + C),绿色 > 0,紫色 < 0)(4 GC 偏度);第五个圆圈显示 GC 含量(5 GC 含量);第六个圆圈显示正链的编码区域和结构 RNA 基因(6 正链);第七个圆圈显示基因结构(7 基因结构);第八个圆圈显示反链的编码区域和结构 RNA 基因(8 反链);(B) Alistipes putredinis 基因组的泛基因组分析。通过 Anvi’o 生成的泛基因组图,基因簇(径向条形图)按基因簇在 Alistipes putredinis 基因组中的频率排序。我们将使用的 15 个 Alistipes putredinis 基因组分为 6 个分支。圆圈从内到外显示,每个基因组中检测到(黑色区域)或未检测到(灰色区域)的基因(圆圈 1-15,6 个分支排列);我们还将基因簇映射到 COG 数据库,并显示 COG 类别(圆圈 16)和已注释的 COG 功能(圆圈 17),这些基因簇中的至少一个基因具有功能注释。在最外圈描述核心基因组和非核心基因组的比例。泛基因组外部展示了显示每个基因组相关属性的条形图。在基因组属性汇总上方,显示了所有百岁组(浅棕色)、老年组(红色)和年轻组(青色)泛基因组样本中基因组的中位数覆盖度(分别以条形图展示)。(C) 使用 fastANI 工具生成的平均核苷酸同一性(ANI)热图,显示对 Alistipes putredinis 各菌株进行多基因组比较得到的基因组相关性。(D) 基于基因簇的 Alistipes putredinis 基因组聚类,及其与系统基因组学方法聚类的比较。顶部的分支树显示了基于泛基因组分析得到的 3958 个基因簇对 15 个 Alistipes putredinis 基因组的聚类。底部的分支树显示了基于40 个核心基因的系统基因组学方法对相同基因组的聚类。颜色表示基于泛基因组分析得到的 6 个系统发育分组。(E) 在非核心基因组比例高的Alistipes putredinis MAGs中识别出的 24 种COG功能。左侧两个面板显示 COG 功能和 COG 类别。右侧的堆叠条形图显示不同系统发育进化支中核心基因组(绿色)和非核心基因组(红色)之间的比例示例。
讨 论
在 EasyMetagenome 中,我们提供了几种处理多种鸟枪法宏基因组数据的选项,适用于不同的场景。针对宏基因组数据中宿主污染的问题,我们开发了 HostPurge(https://github.com/HaoLuo-leo/HostPurge),这是一款集成了流行对齐和 k-mer 方法的新工具,旨在提高大规模数据挖掘的效率、可靠性和可比性。然而,现有工具在处理没有特定宿主参考基因组的短读长高通量测序数据时存在局限性。人工智能算法和第三代长读长测序技术可能有助于克服这一挑战,为建立高质量 MAGs 数据库提供关键支持。这突显了将新算法和专门针对不同宏基因组数据的工作流程整合到EasyMetagenome流程中的迫切需求。
EasyMetagenome 主要在 Linux 系统上运行,充分利用Linux平台对常见宏基因组分析高性能计算工具的强大支持。然而,这一设计可能限制了依赖其他操作系统(如 Windows 或 macOS)用户的可访问性。虽然通过虚拟化或容器化解决方案(如 Docker)可以解决兼容性问题,但这些方法可能会给非专业用户带来额外的复杂性。此外,当处理异常大或复杂的数据集时,流程的可扩展性可能面临挑战。计算兼容性还可能因硬件和软件环境的不同而有所差异,尤其是对于那些高性能计算资源有限的用户。未来版本的 EasyMetagenome 可以探索增强跨平台支持的方案,从而提升其可用性和普及性。
第三代平台生成的测序数据在质量上与第二代测序有所不同,因此需要定制化的分析工具。尽管已经开发了一些用于长读长测序数据的生物信息学软件和方法,如错误校正方法或混合组装校正方法,但针对第三代测序数据的宏基因组分析仍处于初期阶段。基于短读长的算法通常不适用于默认参数下的长读长数据集,这可能导致在解读长测序读长时出现更高的错误率。因此,迫切需要建立一个“金标准”流程来分析第三代宏基因组测序数据,并进一步提高微生物组探索的可比性和可重复性。
未来,EasyMetagenome 将专注于下游数据挖掘,运用新方法深化对宏基因组数据的理解。例如,网络分析已被证明在探索微生物相互作用方面十分有效,通过揭示稳定且功能性强的基因组关系,帮助识别与健康相关的肠道微生物群。近期的“两组核心菌群”(two competing guilds, TCG)模型利用丰度网络分析来识别作为健康指标的核心微生物群特征。另一种方法是广义微生物表型三角化(generalized microbe phenotype triangulation, GMPT),其假设在大多数配对表达中,微生物丰度与疾病严重性呈现强相关(无论是正相关还是负相关),可辅助物种和功能的预测。这一方法突破了传统的差异丰度分析,专注于直接针对与疾病机制相关的核心微生物群体,并已在多项研究中成功应用。随着这些进展的推进,EasyMetagenome 致力于深化我们对微生物功能、相互作用以及与宿主共进化的理解。
结 论
随着对鸟枪法宏基因组数据计算分析可重复性需求的不断增加,建立标准化的生物信息学工作流程变得至关重要。为此,我们开发了EasyMetagenome,这是一款用户友好、灵活的流程,支持全面的数据探索,并在每个步骤提供清晰的参数说明,生成可发布的结果。在本研究中,我们特别展示了两个示例,旨在展示我们流程的灵活性、可用性和可靠性。这一全面的鸟枪法宏基因组分析流程帮助微生物组研究人员专注于解决生物科学中的关键问题,而不必陷入复杂的计算挑战。
展望未来,我们将着重解决影响数据准确性和处理效率的宿主污染问题。我们还将整合针对第三代测序数据优化的工作流程,并引入新兴算法,如深度学习(用于优化微生物组数据读取)、网络分析(用于相互作用研究)和GMPT(用于关键生物标志物识别)。我们的目标是增强微生物组数据分析的可解释性并提高效率。
方 法
EasyMetagenome工作流程概述与软件介绍
我们的流程从Illumina原始测序读长开始,随后进行数据预处理、基于读长的分析、基于组装的分析和分箱。最终的数据将通过R脚本进行统计分析和可视化。
简要来说,质量评估可以使用FastQC(https://www.bioinformatics.babraham.ac.uk/projects/fastqc/)进行。Illumina原始双端序列使用fastp软件进行质量控制。然后,使用KneadData(https://huttenhower.sph.harvard.edu/kneaddata)去除宿主序列,生成清洁的微生物组读长。接着,使用FastQC重新检查清洁读长的质量。物种分类使用Kraken2进行,并估算物种丰度。基因家族和功能通路的相对丰度通过HUMAnN2或HUMAnN3确定。
与此同时,清洁读长使用MEGAHIT或metaSPAdes进行组装。随后,组装的重叠群将通过两种主要途径进行分析。对于基于组装的分析,使用QUAST评估重叠群质量。使用MetaProdigal进行基因预测,通过CD-HIT评估和去除基因冗余,使用Salmon进行基因定量。使用KEGG Ortholog (KO)、碳水化合物活性酶(CAZy)和同源蛋白簇(COG)数据库,利用eggNOG进行微生物组功能注释。抗生素抗性基因也通过ResFams数据库和RGI进行检测。基因的物种注释则使用Kraken2软件。
对于分箱,MAGs通过MetaWRAP中的‘binning’和‘bin_refinement’模块获取。通过dRep处理冗余MAGs,丰度矩阵通过CoverM获取,使用GTDB-tk计算系统发育关系,MAGs的完整性和污染率通过CheckM2估算。MAGs的功能基因注释则再次使用eggnog-mapper、DIAMOND和RGI进行。
EasyMetagenome分析的数据集选择与质量控制
为了展示 EasyMetagenome 的功能,我们对已发布的数据集进行了分析,包括一组人类肠道微生物群样本和一组环境微生物群样本,突出展示了该流程在不同样本类型中的多功能性和可视化能力。对于人类肠道样本,数据集包含三个组别:年轻组(23-47岁)、老年组(85-89岁)和百岁组(100岁以上),每组有六个样本,性别比例相等(表S1)。对于环境样本,我们选择了四个堆肥阶段(冷却、成熟、中温和高温),每个阶段有四个样本(表S2)。这些数据集的详细NCBI序列号可以在表S1和表S2中找到。
所有原始数据都使用 fastp v0.24.0进行了质量控制,随后使用 KneadData v0.12.0 隔离高质量的微生物读长数据,将人类读长数据映射到 GRCh37 人类参考基因组并通过 Bowtie2(v2.5.1;参数:--very-sensitive --dovetail)去除。清洁后的读长数据随后使用 FastQC v0.12.1 进行了质量评估。接着,使用三种互补的方法——基于读长的分析、基于组装的分析和基于分箱的分析——对清洁后的微生物读长数据进行了分析,展示了 EasyMetagenome 流程生成可用于发表的高质量可视化结果的能力。
EasyMetagenome用已发布宏基因组数据基于读长进行分析
在基于读长的分析中,使用高质量的微生物读长进行物种水平的群落谱系分析,利用 MetaPhlAn4 v4.0.6对 mpa_vOct22_CHOCOPhlAnSGB_202212 数据库进行比对,在默认参数下获得所有分类学层级。此外,功能分析通过 HUMAnN3 v3.7进行。随后,应用各种 EasyMetagenome 脚本来可视化这些结果:‘metaphlan_hclust_heatmap.R’ 生成了不同组间各分类学层级分布的热图,‘graphlan_plot54.r’ 则展示了组内整体的分类学结构。此外,‘otu_mean.R’ 计算了组间丰度平均值,‘sp_vennDiagram.sh’ 创建了韦恩图,‘metaphlan_boxplot.R’ 生成了各组不同分类级别的相对丰度箱线图。在 alpha 多样性分析中,使用 ‘otutab_rare.R’ 计算了丰富度、Chao1、ACE、Shannon、Simpson 和Inverse Simpson 等指数,‘alpha_boxplot.R’ 则生成了这些指标的箱线图。应用 ANOVA 和 Tukey 的 HSD 检验来评估差异的显著性并确定 P 值。在 beta 多样性分析中,使用 ‘usearch10’ 计算了 Bray-Curtis、欧氏距离、Jaccard 和曼哈顿距离,并通过 ‘beta_pcoa.R’ 将结果可视化为 PCoA 图,使用 PERMANOVA 和 ADONIS 检验评估统计显著性并计算 P 值。‘tax_stackplot.R’ 脚本用于生成各组之间分类组成的堆叠图,而 ‘compare_stamp.R’ 用于比较两组之间分类丰度差异。此外,‘sp_pheatmap.sh’ 计算了功能通路丰度差异,HUMAnN3 v3.7中的 ‘humann_barplot’ 被用来可视化各组中特定通路的分类学组成。
物种分类使用 Kraken2 v2.1.3进行,采用默认设置和预构建的 PlusPF 数据库,同时使用 Bracken v2.7估计物种丰度,以细化物种级丰度估计。Kraken2 和 MetaPhlAn4 都可以用于分类学谱系分析,但它们采用了不同但互补的方法和参考数据库。Kraken2 使用基于 k-mer 的分类方法,将读长与标准数据库或用户构建的数据库进行比对。相比之下,MetaPhlAn 通过将读长与特定克隆标记基因数据库对齐来进行分类。为了充分利用每种方法的优势,EasyMetagenome 包含了来自两种方法的可视化结果。对于 Kraken2方法获取的数据,我们将其输出格式转换为与 MetaPhlAn 的结构相匹配,并应用上述相同的可视化方法,以生成基于 Kraken2 结果的分类注释结果图。
基于组装的EasyMetagenome宏基因组分析在已发表数据中的应用
在基于组装的分析中,使用 MEGAHIT v1.2.9进行高质量的微生物读长数据的 de novo 组装,随后使用 QUAST v5.0.2评估组装结果。使用 MetaProdigal v2.6.3进行基因预测,组装的重叠群通过 CD-HIT v4.8.1进行聚类,设置 95% 的相似度和 90% 的覆盖度(-aS 0.9 -c 0.95)。接着,使用 Salmon v1.8.0测量每个样本中重叠群的丰度。蛋白功能,包括 KEGG Ortholog (KO)、CAZy和 COGs,基于 eggNOG v5.0 数据集使用 eggNOG-mapper工具进行注释。使用 ‘format_dbcan2list.pl’ 结合 ‘summarizeAbundance.py’生成汇总的丰度表格。随后,使用 R 语言可视化跨四个类别的基因功能注释,按不同层次对注释的功能基因进行分类,并比较不同组之间微生物组的功能模块丰度差异。
使用EasyMetagenome流程对已发布宏基因组数据中的宏基因组组装基因组进行分箱
对于分箱,使用 MEGAHIT v1.2.9组装的重叠群,通过 MetaWRAP v1.3.2中的‘binning’和‘bin_refinement’模块进行 MAGs 分箱。具体而言,使用‘binning’模块 (--metabat2 --maxbin2) 将重叠群聚类为宏基因组 bin,然后使用‘bin_refinement’模块 (-c 50 -x 10) 进行精细化。之后,使用 dRep v2.6.2对精细化后的 bins 进行去重处理,参数设置为‘-sa 0.95 -nc 0.3 -p 16 -comp 50 -con 10’。接着,使用 CoverM v0.6.1 获得去重基因组在各个样本中的丰度矩阵。
精细化 bins 的分类通过使用 GTDB-tk v2.4.0 (r214) 数据库中的‘gtdbtk classify_wf’模块进行,并通过‘gydbtk infer’模块推断它们的系统发育关系。使用 CheckM2 v1.0.1估算 MAGs 的完整性和污染率,依据以下阈值选择高质量 MAGs:高质量:≥ 90% 完整性和 ≤ 5% 污染率(对于某些环境样本,可能需要根据实际情况进行调整);中等质量:> 50% 完整性和 ≤ 5% 污染率;低质量:< 50% 完整性。随后,使用 eggnog-mapper v2.1.6、DIAMOND v2.0.13和 RGI v5.2.0对高质量 MAGs 进行功能注释,功能类别包括 KEGG Ortholog (KO)、CAZy 和 COGs。最后,使用 R语言可视化 MAGs 的基因组质量,进行系统发育分析,并展示 MAGs 的分布及其功能基因注释结果。
Alistipes putredinis的基因组结构与泛基因组分析
在获得所有样本的MAG注释结果后,我们从人类肠道宏基因组数据中选择了Alistipes putredinis(Alistipes putredinis MAG)进行单菌基因组注释和泛基因组分析,因为其具有较高的完整性(96.23%)和较低的污染率(0.67%)。对于单菌基因组注释,我们首先使用Bakta v1.9.4对Alistipes putredinis MAG进行注释。获得基因组注释结果后,我们将注释文件导入Proksee软件进行进一步分析和可视化。我们还计算了GC偏度,以评估所选基因组中鸟嘌呤(G)和胞嘧啶(C)的相对含量,并使用mobileOG-db进行Alistipes putredinis的其他基因组注释,以识别移动直系同源群。
对于泛基因组分析,我们使用anvi’o v8对Alistipes putredinis的组装基因组进行分析,结合了Alistipes putredinis MAG和14个来自NCBI的公开可用Alistipes putredinis分离株(表S3)。这15个Alistipes putredinis基因组用于通过anvi’o v8中的“anvi-gen-重叠群-database”功能构建重叠群数据库。随后,使用“anvi-run-hmms”功能提取细菌单拷贝基因信息,并使用“anvi-run-ncbi-cogs”功能获得NCBI COG功能注释。我们将来自人类肠道微生物群样本的短读序列映射到构建的重叠群数据库,并分别使用Bowtie2和samtools对招募到的读长进行排序。通过从不同年龄组招募宏基因组短读序列后,我们使用“anvi-profile”功能处理招募文件,并生成包含重叠群数据库的基因覆盖和检测信息的profile数据库。这些profile被合并为每个样本的单一数据库,通过“anvi-import-collection”功能将重叠群重叠群与其各自的基因组连接起来。最后,我们使用“anvi-gen-genomes-storage”功能进行基因组存储,并使用“anvi-pan-pangenome”功能生成Alistipes putredinis的泛基因组。随后,使用anvi’o交互界面中的“anvi-display-pan”功能可视化泛基因组。
在获得Alistipes putredinis的泛基因组后,我们将基于泛基因组分析中检测到的所有基因簇进行的Alistipes putredinis基因组聚类与基于系统基因组学的聚类进行了比较,后者是使用从1,322个核心基因中筛选出的40个核心基因进行的。为了进一步评估基因组相似性,我们使用fastANI v1.34计算了Alistipes putredinis MAG与其他14个已发布的Alistipes putredinis基因组之间的平均核苷酸相似度(ANI)。此外,为了检查Alistipes putredinis泛基因组的功能组成,我们展示了通过COG数据库识别的所有Alistipes putredinis基因组中具有异常高AG(accessory genomes)比率的基因。
代码和数据可用性
EasyMetagenome 可免费使用,以 Shell 脚本和 R语言实现,易于安装。详细的分析步骤在 GitHub https://github.com/YongxinLiu/EasyMetagenome 上提供。所有图形数据和补充表格都可通过 GitHub https://github.com/YongxinLiu/EasyMetagenome/tree/master/appendix 访问。更详细的数据信息可联系通讯作者获取。补充材料(文本、图、表、中文翻译版本或视频)也可从线上(http://www.imeta.science/)获取。
引文格式:
Defeng Bai, Tong Chen, Jiani Xun, Chuang Ma, Hao Luo, Haifei Yang, et al. 2025. “EasyMetagenome: A user-friendly and flexible pipeline for shotgun metagenomic analysis in microbiome research.” iMeta 4: e70001. https://doi.org/10.1002/imt2.70001.
作者简介

白德凤(第一作者)
● 中国农业科学院深圳农业基因组所博士后。
● 博士毕业于中国科学院动物研究所。目前研究方向为宏基因组数据分析及挖掘,肠道微生物和疾病关系研究。相关学术成果已发表于iMeta, Landscape Ecology, Zoological Research等期刊。

陈同(第一/通讯)
● 中国中医科学院副研究员,道地药材品质保障与资源持续利用全国重点实验室青年 PI,iMeta期刊执行主编,生信宝典公众号创始人。
● 主要研究方向是中药资源大数据+人工智能驱动的高质量中药材的鉴定、遗传改造、生态重塑、异源合成和中药新资源的发现等,在Cell Stem Cell (封面文章),Nucleic Acids Research,Nature communications, Protein & Cell, iMeta等高水平杂志以第一或通讯作者发表文章十余篇,累积引用 3000 +次;开发在线绘图和分析平台 ImageGP、BIC、EVenn、植物整合基因组平台IMP (获中华中医药学会 2023 年年度十大学术进展之一),使用超 70 万人次;运营有十五万人关注的微信公众号《生信宝典》,分享有1400 多篇生物信息分析原创文章、教程和视频,阅读播放千万次。联合创办iMeta期刊,现为执行主编,致力于打造微生物和生物信息领域的国产高水平综合性杂志。

荀佳妮(第一作者)
● 中国农业科学院深圳农业基因组所生物信息学硕士在读,本科毕业于华北理工大学生命科学学院。
● 主要研究方向为宏基因组数据分析和挖掘。

马闯(第一作者)
● 中国农业科学院深圳农业基因组所客座硕士,安徽农业大学园艺学院研三在读。
● 主要研究方向为百岁老人肠道病毒组、土壤微生物组。

罗豪(第一作者)
● 中国农业科学院深圳农业基因组研究所,在读博士研究生。
● 研究方向为人工智能在微生物组数据分析中的应用,在iMeta、iMetaOmics、GigaScience、Journal of Hazardous Materials等期刊发表论文5篇,其中第一/共同第一作者3篇。

杨海飞(第一作者)
● 青岛农业大学生命科学院在读硕士研究生。
● 主要研究方向为食物微生物组、宏基因组。

文涛(通讯作者)
● 博士,南京农业大学钟山青年研究员, iMeta 期刊青年编委,“微生信生物”公众号创始人。
● 研究方向为根际微生物生态,擅长使用多组学解析土壤微生物群落过程,开发了ggClusterNet, EasyStat等R包, EasyAmplicon、EasyMetabolome等组学分析流程。以第一作者在iMeta、Microbiome、ISME J、Fundamental Research、 Horticulture Research、SEL、BMC plant biology等期刊发表论文10余篇。

陈实富(通讯作者)
● 中科院博士,海普洛斯创始人兼首席技术官,中科院深圳先进技术研究院客座研究员。
● 2019年深圳市青年科技奖获得者,2021深圳市科技达人,2022广东省创新达人,以项目负责人获2022深圳科技进步奖获。开源项目组OpenGene的发起人,多款热门生物信息学软件的作者。发表国际期刊和会议论文60余篇,其中一作兼通讯最高单篇引用超过16000次(近五年中国生物学者单篇引用前三)。申请30余项发明专利和40多项软件著作权,是中国抗癌协会、中国临床肿瘤学会以及美国肿瘤学会会员,中国抗癌协会肿瘤标志专委会青年委员,肿瘤测序及大数据分析专委会委员,中国工业与应用数学学会“数学与产业专业委员会”委员。

李国梁(通讯作者)
● 中国科学院动物研究所博士,江西农业大学林学院青年教授。
● 研究方向为鸟类生态学,主要关注鸟类迁徙机制、鄱阳湖环境污染、鸟类群落组装机制等科学问题。

高云云(通讯作者)
● 中国农业科学院深圳农业基因组研究所博士后,2022年博士毕业于北京林业大学。
● 目前研究方向为宏基因组方法开发,相关成果已发表于iMeta、Protein & Cell等期刊。

刘永鑫(通讯作者)
● 中国农业科学院深圳农业基因组研究所研究员,微生物组与营养健康团队首席,博士生导师。iMeta期刊执行主编,宏基因组公众号创始人,中国微生物学会微生物组专委会委员。
● 研究方向聚焦微生物组学方法开发、功能挖掘和科学传播,以第一或通讯作者(含共同) 在Nature Biotechnology、Nature Microbiology 等发表研究论文40余篇,在Current Opinion in Microbiology、Protein Cell等撰写综述20余篇,合作在Science、Cell Host & Microbe等期刊发表论文20余篇,累计发文80余篇,被引用23000余次,连续入选全球前2%顶尖科学家榜单。主持国自然青年/面上,中国科学院/中国农科院/深圳市等人才项目。创办宏基因组公众号,17万+同行关注,累计阅读量超6千万。主编《微生物组实验手册》专著,联合152家单位的352位同行参与,共同打造本领域长期更新的中文百科全书。发起iMeta期刊并任执行主编,影响因子33.2位列微生物学科研究类期刊全球第一。为Cell Host & Microbe、Nature Communications、Microbiome、ISME、NAR等期刊审稿300余次。
更多推荐
(▼ 点击跳转)
iMeta | 引用20000+,海普洛斯陈实富发布新版fastp,更快更好地处理FASTQ数据
iMeta | 兰大张东组:使用PhyloSuite进行分子系统发育及系统发育树的统计分析
iMeta | 唐海宝/张兴坦-用于比较基因组学分析的多功能分析套件JCVI
iMeta封面

1卷1期

1卷2期

1卷3期

1卷4期

2卷1期

2卷2期

2卷3期

2卷4期

3卷1期

3卷2期

3卷3期

3卷4期

3卷5期

3卷6期

4卷1期

4卷2期

4卷3期

4卷4期

4卷5期

4卷6期
iMetaOmics封面

1卷1期

1卷2期

2卷1期

2卷2期

2卷3期

2卷4期
iMetaMed封面

1卷1期

1卷2期
期刊简介
“iMeta” 是由威立、宏科学和本领域数千名华人科学家合作出版的开放获取期刊,主编由中科院微生物所刘双江研究员和荷兰格罗宁根大学傅静远教授担任。目的是发表所有领域高影响力的研究、方法和综述,重点关注微生物组、生物信息、大数据和多组学等前沿交叉学科。目标是发表前10%(IF > 20)的高影响力论文。期刊特色包括中英双语图文、双语视频、可重复分析、图片打磨、60万用户的社交媒体宣传等。2022年2月正式创刊!相继被Google Scholar、PubMed、SCIE、ESI、DOAJ、Scopus等数据库收录!2025年6月影响因子33.2,中科院分区生物学1区Top,位列全球SCI期刊前千分之三(65/22249),微生物学科2/163,仅低于Nature Reviews,学科研究类期刊全球第一,中国大陆5/585!
“iMetaOmics” 是“iMeta” 子刊,主编由中国科学院北京生命科学研究院赵方庆研究员和香港中文大学于君教授担任,目标是成为影响因子大于10的高水平综合期刊,欢迎投稿!
"iMetaMed" 是“iMeta” 子刊,专注于医学、健康和生物技术领域,目标是成为影响因子大于15的医学综合类期刊,欢迎投稿!
iMeta主页:
http://www.imeta.science
姊妹刊iMetaOmics主页:
http://www.imeta.science/imetaomics/
出版社iMeta主页:
https://onlinelibrary.wiley.com/journal/2770596x
出版社iMetaOmics主页:
https://onlinelibrary.wiley.com/journal/29969514
出版社iMetaMed主页:
https://onlinelibrary.wiley.com/journal/3066988x
iMeta投稿:
https://wiley.atyponrex.com/journal/IMT2
iMetaOmics投稿:
https://wiley.atyponrex.com/journal/IMO2
iMetaMed投稿:
https://wiley.atyponrex.com/submission/dashboard?siteName=IMM3
邮箱:
office@imeta.science

腾讯云面向开发者汇聚海量精品云计算使用和开发经验,营造开放的云计算技术生态圈。
更多推荐
 0
0 0
0- 0



 已为社区贡献55条内容
已为社区贡献55条内容

所有评论(0)