降解组数据分析笔记
降解组数据=被切开的mRNA碎片测序数据 (Degradome sequencing / PARE sequencing Parallel Analysis of RNA Ends)。**只测 mRNA 被切开后的 5’ 末端片段**, 用来**精准找到 miRNA 在 mRNA 上的剪切位点**。
降解组数据
降解组数据=被切开的mRNA碎片测序数据 (Degradome sequencing / PARE sequencing Parallel Analysis of RNA Ends)。
**只测 mRNA 被切开后的 5’ 末端片段**, 用来**精准找到 miRNA 在 mRNA 上的剪切位点**。
1、它是怎么来的?
1. 细胞里有很多 mRNA
2. **miRNA 引导 AGO 蛋白**,在特定位置把 mRNA 切断
3. 被切断的 mRNA 会留下一个**新的 5’ 端**
4. 降解组测序 **只富集、只测序这些新 5’ 端碎片** 所以: **降解组数据 = 一堆来自“被切开位置”的短序列 reads
2、降解组数据本质是什么?
1. **序列信息**:这段碎片来自哪段 mRNA
2. **位置计数**:在 mRNA 的**哪个碱基位置**被切开、切了多少次 (可以把它理解成: 一张“剪切位点热力图”** 哪里切得多,reads 就多;哪里是 miRNA 真实靶标,就会出现尖锐的峰。)
3、数据格式长什么样?
降解组分析核心文件格式详解表:
| 阶段 | 格式后缀 | 全称 / 类型 | 核心作用 | 关键特点(必看) | 对应你图片中的描述 |
|---|---|---|---|---|---|
| 原始数据 | .fastq |
高通量测序原始格式 | 存储测序仪下机的原始读段(Reads) | 包含序列信息 + 质量值;是所有分析的起点 | ✅ 原始测序 reads |
| 比对结果 | .bam |
二进制比对映射格式 | 存储 Reads 比对到基因组 / 转录组的位置信息 | 由 SAM 格式压缩而来,体积小、便于索引;核心关注 5’端坐标 | ✅ 比对到基因组 / 转录组的文件 |
| 信号密度 | .bed |
浏览器可展示数据格式 | 标注剪切位点位置及覆盖度 | 文本格式,列数固定,可直接导入 IGV 查看峰位 | ✅ 剪切位点信号密度(最常用) |
| 信号密度 | .wig/.bw |
基因组信号轨道格式 | 展示全基因组剪切信号的连续密度 | 适合画全转录本的覆盖趋势;.bw是其二进制压缩版 |
✅ 剪切位点信号密度(最常用) |
| 可视化结果 | T-plot | 靶基因剪切位点图谱 | 直观验证 miRNA 靶标 | 不是文件格式,是由 bed/wig 数据生成的图表;峰尖对应真实剪切位点 | ❌ (是图表,非文件格式) |
4、和普通转录组、小RNA组的区别
**转录组**:测所有mRNA,看表达量
- **小RNA组**:测miRNA等小RNA
- **降解组**:**只测被切开的mRNA末端,找剪切位点**
一句话总结:**降解组 = miRNA 剪切 mRNA 的“现场证据”数据**
5、它能解决什么问题?
1. 找 **miRNA 的真实靶基因** 2. 精确定位 **剪切位点(10~11位之间)** 3. 验证预测的 miRNA–mRNA 对是否真实 4. 画 T-plot 提供文章里最硬核的证据
数据分析
一、无参转录组组装与预处理
选取无参转录本:
二、降解组数据预处理
质量过滤的核心目标是:去除低质量、接头污染、冗余和非目标 RNA 的 reads,保留高可信度的 mRNA 降解片段。
完整的降解组质控应该包含哪些核心指标:
| 质控模块 | 核心指标 | 合格标准参考 |
|---|---|---|
| 原始数据质量 | Q20、Q30 比例 | Q30 > 80%,Q20 > 95% |
| 数据过滤 | clean reads 比例、接头残留率 | 接头残留率 < 0.1% |
| 污染评估 | rRNA/tRNA 占比 | rRNA reads < 10%(视物种而定) |
| 比对分析 | 比对率、唯一比对率 | 比对率 > 70%,唯一比对率 > 60% |
| 序列特征 | GC 含量分布、重复率 | GC 含量符合物种预期,无明显峰 |
质控
1.单碱基质量分布:
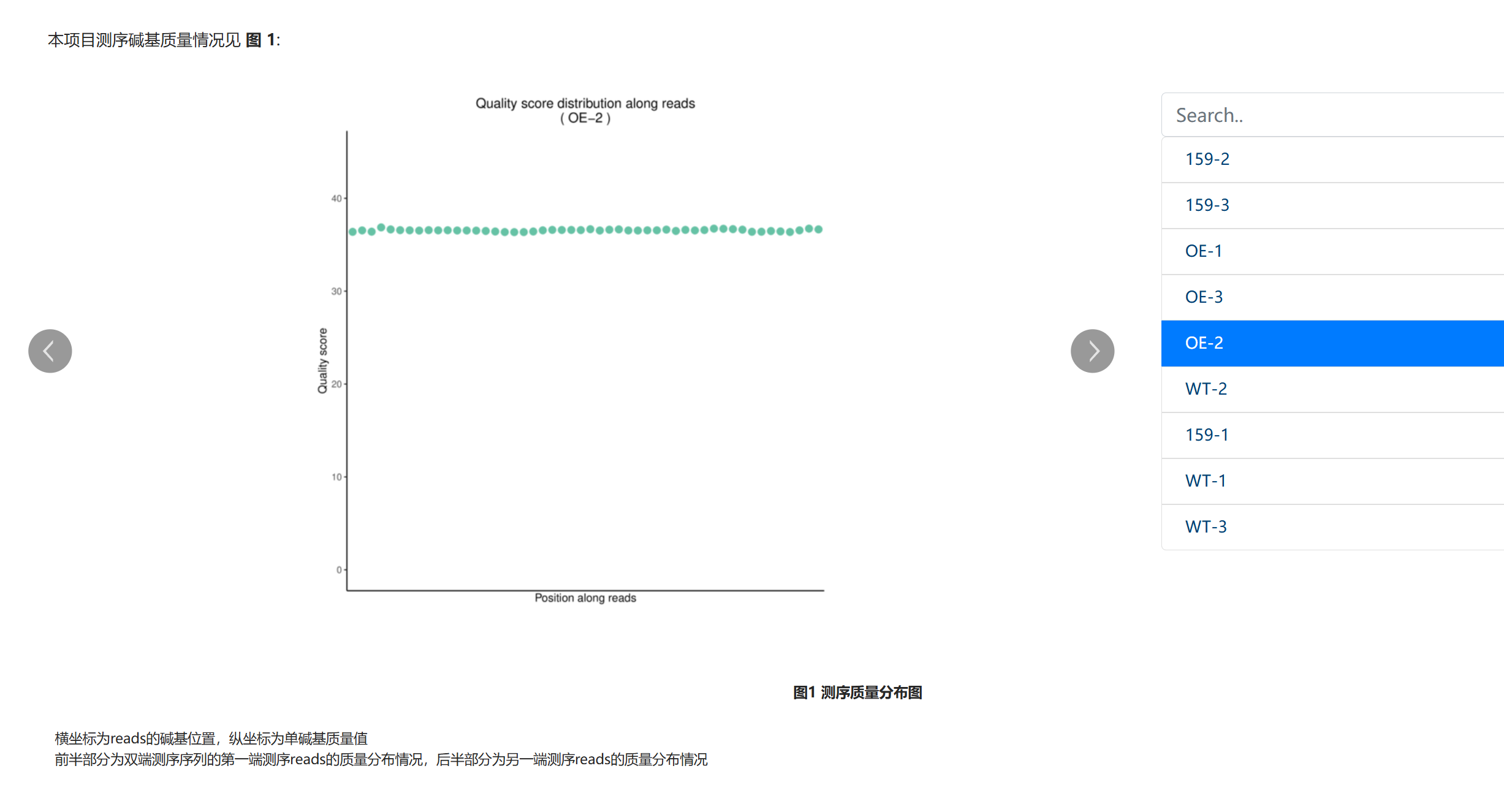
公司提供的测序质量分布图:
-
纵轴代表单碱基质量值(Quality score,Q 值),图中所有位点的 Q 值均稳定在36~38 左右。
-
测序行业通用标准:Q≥20(碱基识别准确率 99%)、Q≥30(准确率 99.9%,即 “Q30”)。
-
该样本全读长范围的 Q 值远高于 Q30,意味着碱基识别错误率极低(远低于 0.1%),原始序列的准确性有极强保障。
-
-
质量值的均匀性横轴为 reads 的碱基位置,图中绿色点(各位置质量值)几乎呈一条平直的水平线,无明显的 “两端下降” 或 “中间波动”。
-
降解组测序对5’端碱基的准确性要求极高(核心是定位 5’端剪切位点),该样本全位置质量均匀,避免了因末端质量差导致的剪切位点定位偏差,是后续比对和位点鉴定的重要前提。
-
2.原始数据质控评估(FastQC + MultiQC)
用 FastQC 生成单样本报告:
fastqc OE-2_raw.fastq -o qc_reports/用 MultiQC 汇总所有样本,方便对比:
multiqc qc_reports/ -o multiqc_report/重点关注:
- 碱基质量分布(Q20/Q30 比例)
- 接头污染情况
- GC 含量分布
- 重复序列比例
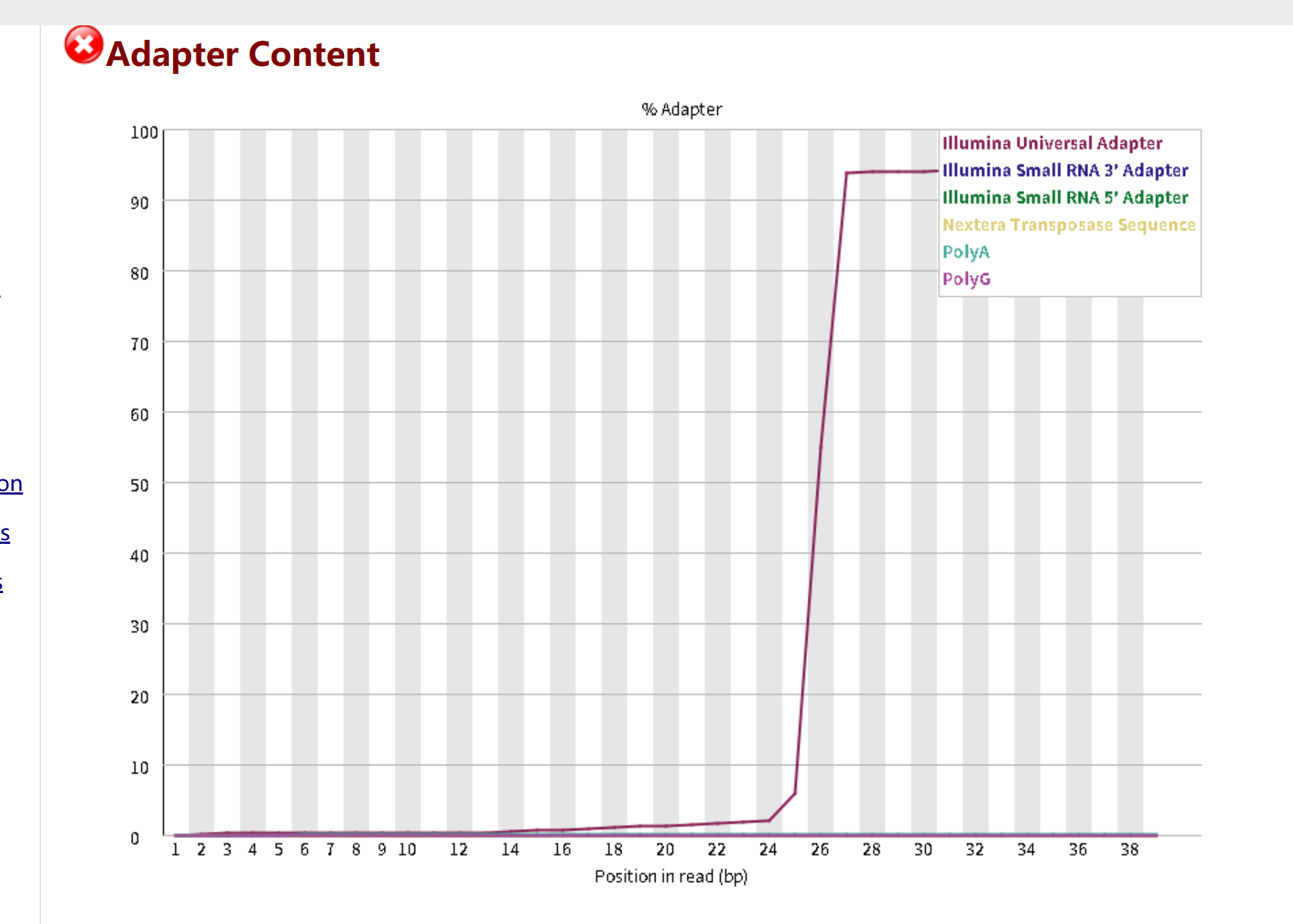
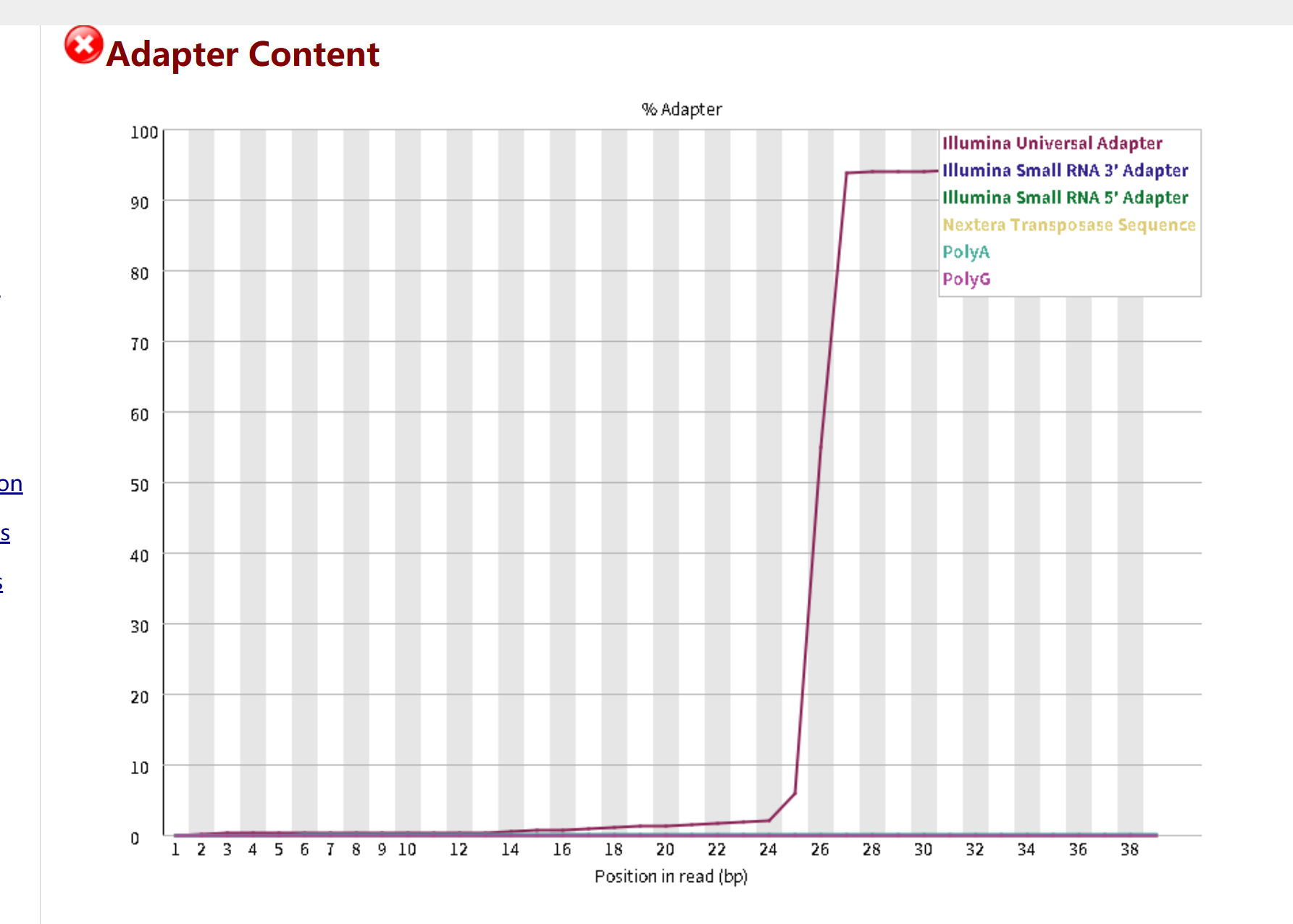
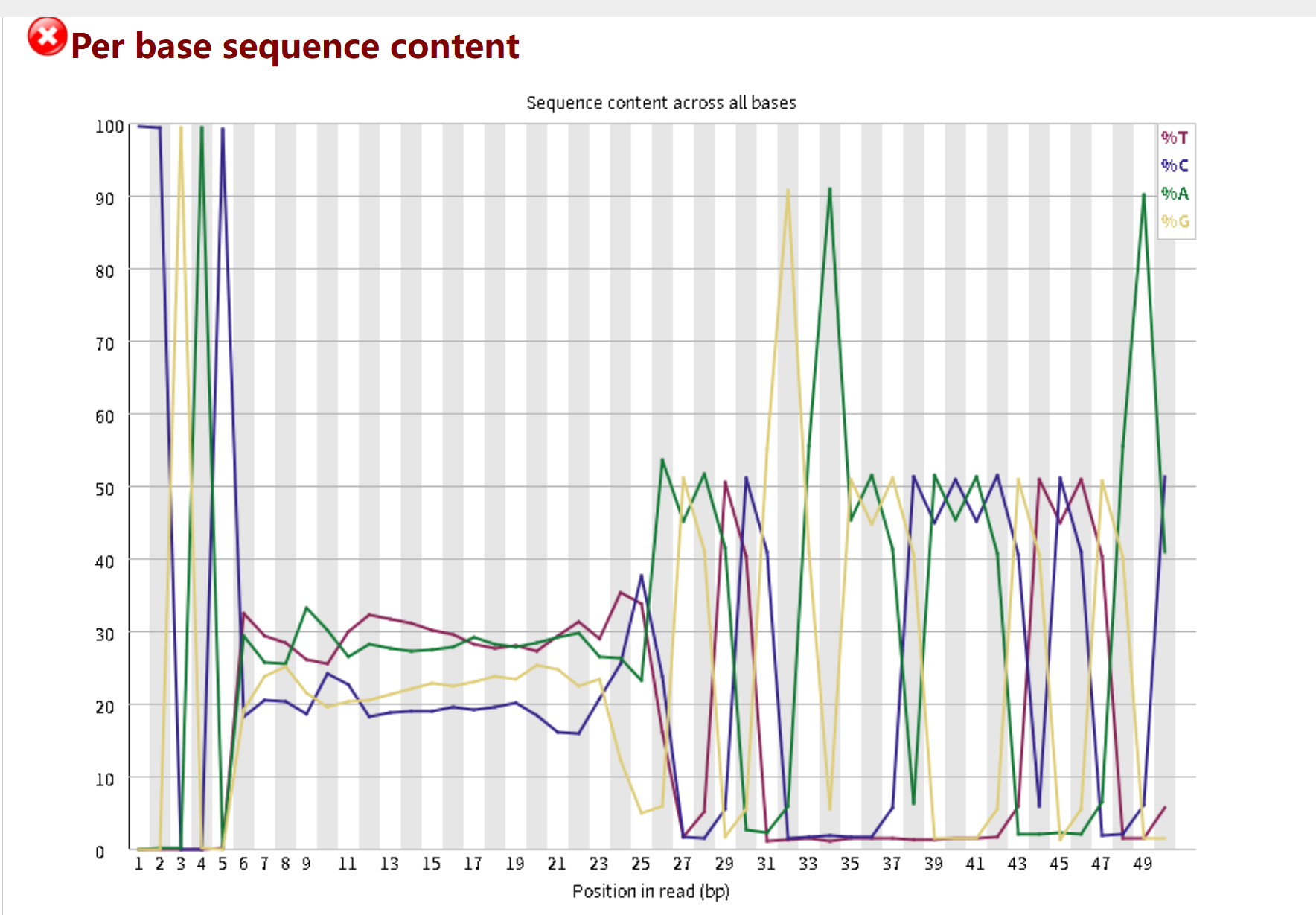
Adapter Content(接头污染)
-
现象:在 reads 的 26 bp 之后,
Illumina Small RNA 3' Adapter的比例急剧升高,接近 100%。 -
解读:这是典型的小 RNA / 降解组测序接头残留。因为降解组 reads 本身很短(通常 36-50 nt),当插入片段短于测序读长时,测序仪会直接读到 3' 端的接头序列。
-
影响:接头序列会严重干扰后续比对,导致大量 reads 无法正确定位到转录组。
-
处理建议:必须在过滤步骤中明确加入该接头序列进行修剪。在 Cutadapt 中使用
-a AGATCGGAAGAGCACACGTCTGAACTCCAGTCAC来去除它。
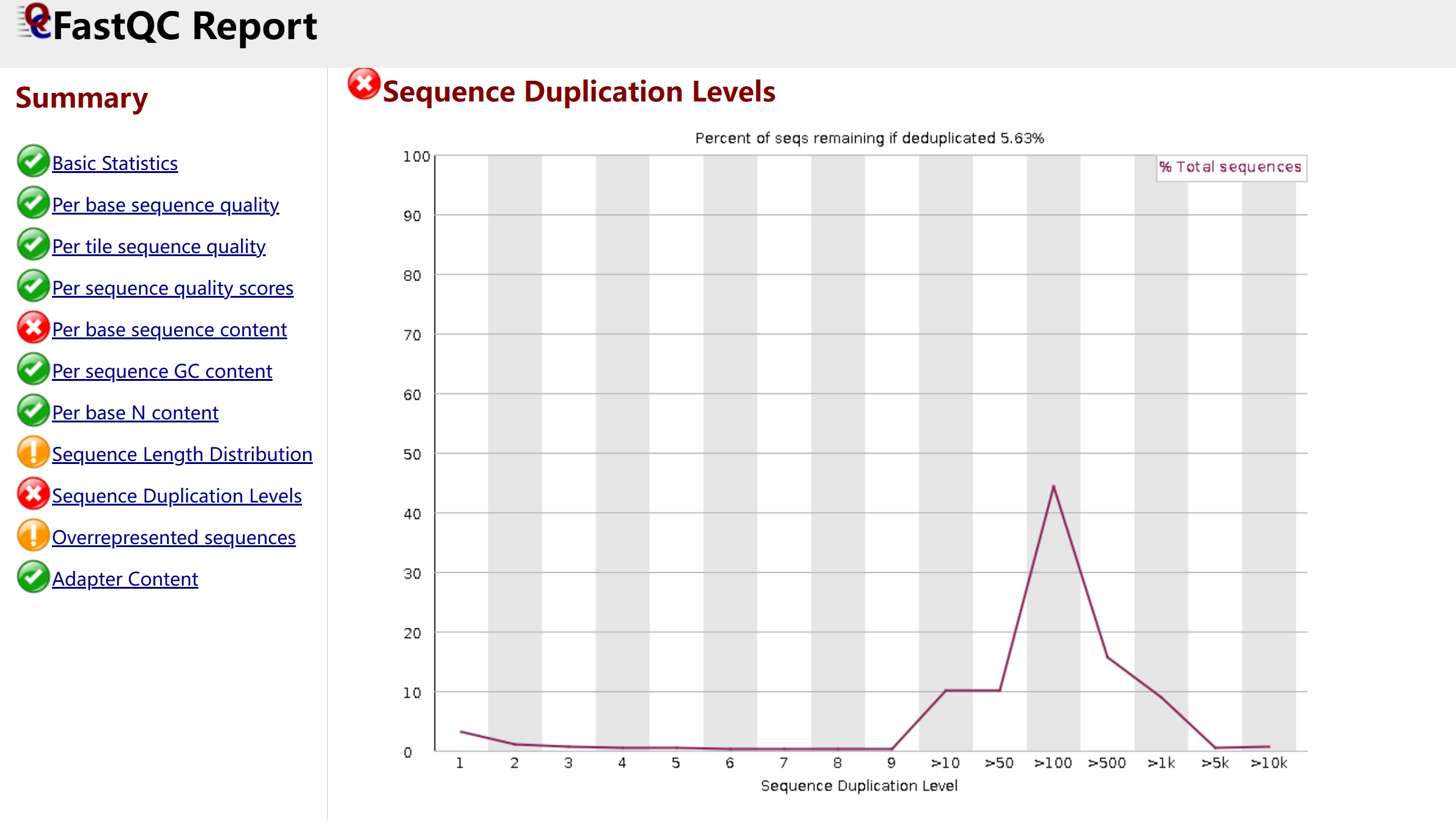
2. Sequence Duplication Levels(序列重复水平)
-
现象:大部分序列的重复次数很低,但在重复次数 > 100 的区间出现了一个明显的尖峰,去重后仅剩 7.93% 的序列保留。
-
解读:这在降解组 / 小 RNA 测序中非常常见。高重复率主要来自于:
-
真实的生物学信号:同一剪切位点被多次测序,产生大量相同 reads。
-
PCR 扩增偏好:某些序列在扩增中被过度复制。
-
-
影响:高重复率会导致计算量增大,并可能掩盖真实的低丰度剪切位点。
-
处理建议:
-
必须进行 ** 去冗余(collapsing)** 处理,将完全相同的 reads 合并为一条,并记录其出现次数。
-
后续分析时,使用去重后的数据,同时保留计数信息,用于剪切位点的定量。
-
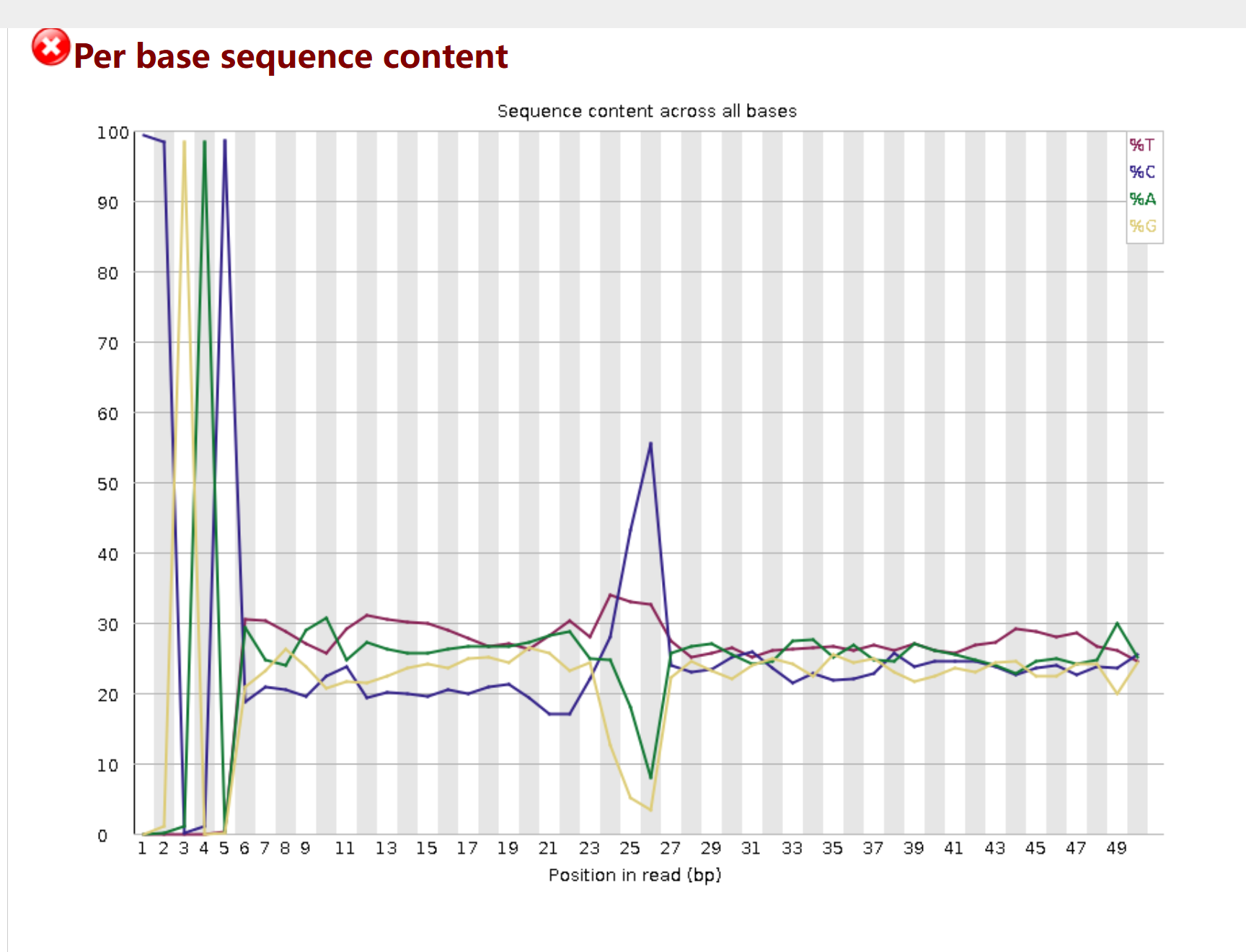
3. Per base sequence content(每碱基序列组成)
-
现象:在 reads 的前 5 个碱基位置,四种碱基(A/T/C/G)的比例严重失衡,出现极端峰值;在 reads 中部(如 25-35 bp)也有明显波动。
-
解读:
-
前 5 bp 的偏倚:这是降解组 / PARE 测序的典型特征,源于 5' 端接头连接和 RNA 酶切产物的富集,属于技术偏倚,而非生物学问题。
-
中部波动:可能是接头残留或低质量碱基导致的序列组成异常,也与降解组短读长的特性有关。
-
-
影响:这种偏倚会影响序列比对的准确性,尤其是在使用依赖碱基组成的比对算法时。
-
处理建议:
-
可以考虑在修剪接头和低质量碱基后,再检查碱基组成是否恢复正常。
-
如果前几 bp 的偏倚依然严重,可以在比对前将 reads 的前几个碱基(如前 4-6 bp)进行修剪,以提高比对率。
-
#!/bin/bash
# 批量解压当前目录及子目录下的所有 .fq.gz 文件
# 定义根目录
ROOT_DIR="./"
# 遍历根目录下所有子目录,查找 .fq.gz 文件并解压
find ${ROOT_DIR} -type f -name "*.fq.gz" | while read gz_file; do
# 获取压缩包所在的目录
target_dir=$(dirname "${gz_file}")
echo "====================================="
echo "开始解压:${gz_file}"
echo "解压到目录:${target_dir}"
# 进入目标目录执行解压,避免文件跑到别的地方
cd "${target_dir}" || exit
gunzip -q "$(basename "${gz_file}")"
# 解压成功/失败提示
if [ $? -eq 0 ]; then
echo "✅ 解压完成:${gz_file}"
else
echo "❌ 解压失败:${gz_file}"
fi
# 回到脚本初始目录
cd - > /dev/null || exit
done
echo "====================================="
echo "📌 批量解压任务执行完毕!"# 基础版:仅去除接头+质量过滤(推荐首选)
cutadapt \
-a TGGAATTCTCGGGTGCCAAGG \ # Illumina Small RNA 3' Adapter核心序列(适配你的质控图)
-a A{10} \ # 去除连续≥10个A的polyA尾
-q 20,20 \ # 5'和3'端质量阈值:低于Q20的碱基修剪
-m 18 \ # 保留长度≥18nt的reads(降解组有效片段最小长度)
-M 50 \ # 丢弃长度>50nt的reads(过滤异常长片段)
--trim-n \ # 去除reads中的N碱基
-o OE-2_trimmed.fastq \ # 输出去接头后的文件
OE-2_raw.fastq # 输入原始fastq文件查找 cutadapt 所在的环境:
# 遍历所有环境,检查cutadapt是否存在
for env in $(mamba env list | grep -v "^#" | awk '{print $1}'); do
echo "检查环境: $env"
mamba run -n $env which multiqc 2>/dev/null
done#!/bin/bash
# 批量对目录下的fq文件执行cutadapt去接头+质量过滤
# 适配规则:原始文件 *.fq → 输出文件 *_cutadapt.fq
# ===================== 可配置参数(根据你的需求修改)=====================
ROOT_DIR="./" # 要处理的根目录(当前目录或绝对路径)
ADAPTER_SEQ="TGGAATTCTCGGGTGCCAAGG" # Illumina Small RNA 3' Adapter核心序列
POLYA_SEQ="A{10}" # polyA尾过滤规则(连续≥10个A)
QUAL_THRESH="20,20" # 5'/3'端质量阈值
MIN_LEN=18 # 保留最小长度(nt)
MAX_LEN=50 # 丢弃最大长度(nt)
# =========================================================================
echo "===== 开始批量执行cutadapt质控 ====="
echo "处理目录:${ROOT_DIR}"
echo "适配规则:${ADAPTER_SEQ} | polyA:${POLYA_SEQ} | 质量:${QUAL_THRESH} | 长度:${MIN_LEN}-${MAX_LEN}nt"
echo "文件匹配:查找所有 .fq 后缀文件,输出为 *_cutadapt.fq"
echo "====================================="
# 遍历根目录下所有.fq文件(递归查找子目录,排除已处理的_cutadapt.fq)
find ${ROOT_DIR} -type f -name "*.fq" ! -name "*_cutadapt.fq" | while read raw_fq; do
# 1. 获取文件基本信息
file_dir=$(dirname "${raw_fq}") # 文件所在目录
file_name=$(basename "${raw_fq}") # 文件名(如OE-2.fq)
# 替换输出文件名:把.fq改成_cutadapt.fq(例:OE-2.fq → OE-2_cutadapt.fq)
trimmed_fq="${file_dir}/${file_name%.fq}_cutadapt.fq"
echo "-------------------------------------"
echo "正在处理:${raw_fq}"
echo "输出文件:${trimmed_fq}"
# 2. 执行cutadapt命令 集群环境下不能出现反斜杠
mamba run -n cutadapt cutadapt \
-a "${ADAPTER_SEQ}" \ # 3'接头序列
-a "${POLYA_SEQ}" \ # polyA尾过滤
-q "${QUAL_THRESH}" \ # 质量过滤
-m "${MIN_LEN}" \ # 最小长度
-M "${MAX_LEN}" \ # 最大长度
--trim-n \ # 去除N碱基
-o "${trimmed_fq}" \ # 输出文件
"${raw_fq}" # 输入文件
# 3. 检测执行结果
if [ $? -eq 0 ]; then
echo "✅ 处理成功:${file_name}"
else
echo "❌ 处理失败:${file_name}(请检查文件或参数)"
fi
done
echo "====================================="
echo "📌 批量cutadapt质控任务执行完毕!"
echo "✅ 成功文件可查看 *_cutadapt.fq;❌ 失败文件请检查日志提示。"#!/bin/bash
# 批量对目录下的fq/fastq文件执行FastQC质控,并使用MultiQC汇总报告
# 结果输出:新建的qc_reports文件夹(包含单个FastQC报告+MultiQC汇总报告)
# ===================== 可配置参数(根据你的需求修改)=====================
ROOT_DIR="./" # 要处理的根目录(当前目录或绝对路径)
QC_OUTPUT_DIR="./qc_reports" # 质控报告汇总文件夹
FILE_PATTERN="*.fq" # 要检测的文件后缀(支持*.fq/*.fastq/*.fq.gz/*.fastq.gz)
THREADS=8 # FastQC使用的线程数(根据集群资源调整)
# =========================================================================
# 创建质控报告目录(如果不存在)
mkdir -p "${QC_OUTPUT_DIR}"
echo "===== 开始批量执行FastQC + MultiQC质控 ====="
echo "处理目录:${ROOT_DIR}"
echo "文件匹配:${FILE_PATTERN}"
echo "报告输出:${QC_OUTPUT_DIR}"
echo "使用线程:${THREADS}"
echo "====================================="
# 第一步:批量执行FastQC(递归查找目标文件,排除已处理的cutadapt文件避免重复)
find ${ROOT_DIR} -type f -name "${FILE_PATTERN}" ! -name "*_cutadapt.fq" | while read qc_file; do
echo "-------------------------------------"
echo "正在对文件执行FastQC:${qc_file}"
# 集群环境下执行FastQC(通过mamba激活环境,无反斜杠换行)
mamba run -n fastqc fastqc \
-t "${THREADS}" \
-o "${QC_OUTPUT_DIR}" \
"${qc_file}"
# 检测FastQC执行结果
if [ $? -eq 0 ]; then
echo "✅ FastQC处理成功:${qc_file}"
else
echo "❌ FastQC处理失败:${qc_file}(请检查文件或集群环境)"
fi
done
# 第二步:使用MultiQC汇总所有FastQC报告
echo "-------------------------------------"
echo "开始执行MultiQC汇总报告..."
mamba run -n multiqc multiqc \
"${QC_OUTPUT_DIR}" \
-o "${QC_OUTPUT_DIR}" \
--title "FastQC_MultiQC_Report"
# 检测MultiQC执行结果
if [ $? -eq 0 ]; then
echo "✅ MultiQC汇总成功!汇总报告已保存至:${QC_OUTPUT_DIR}/multiqc_report.html"
else
echo "❌ MultiQC汇总失败(请检查FastQC报告是否生成)"
fi
echo "====================================="
echo "📌 批量FastQC+MultiQC质控任务执行完毕!"
echo "✅ 单个FastQC报告:${QC_OUTPUT_DIR}(*.html/*.zip)"
echo "✅ 汇总报告:${QC_OUTPUT_DIR}/multiqc_report.html"#在powershell中输入,传输文件
#在 scp 命令中加上 -r 参数,表示递归传输整个目录:
#绝对路径根目录pwd
scp [集群用户名]@[集群服务器IP]:[集群文件的绝对路径] [本地电脑的保存路径]#直接在原始数据里找 adapter,判断这个接头是否正确
grep -c TGGAATTCTCGGGTGCCAAGG 159-1.fqfastqc_top_overrepresented_sequences_table这个文件:
Sample Reports Occurrences % of all reads CCAGATCGGAAGAGCACACGTCTGAACTCCAGTCACACTGAGCGATCGCG 1 35468 0.012459432678414411
overrepresented 序列都包含这一段:
AGATCGGAAGAGCACACGTCTGAACTCCAGTCAC
这就是标准 Illumina TruSeq 3' adapter 核心序列。
而不是:
TGGAATTCTCGGGTGCCAAGG
所以你之前 trim 的 adapter 完全用错了。
这就是为什么:
-
FastQC 显示大量 adapter
-
cutadapt 只识别 1.8%
因为你 trim 的根本不是它。
修改后:

接头去除干净后,还是有些问题:
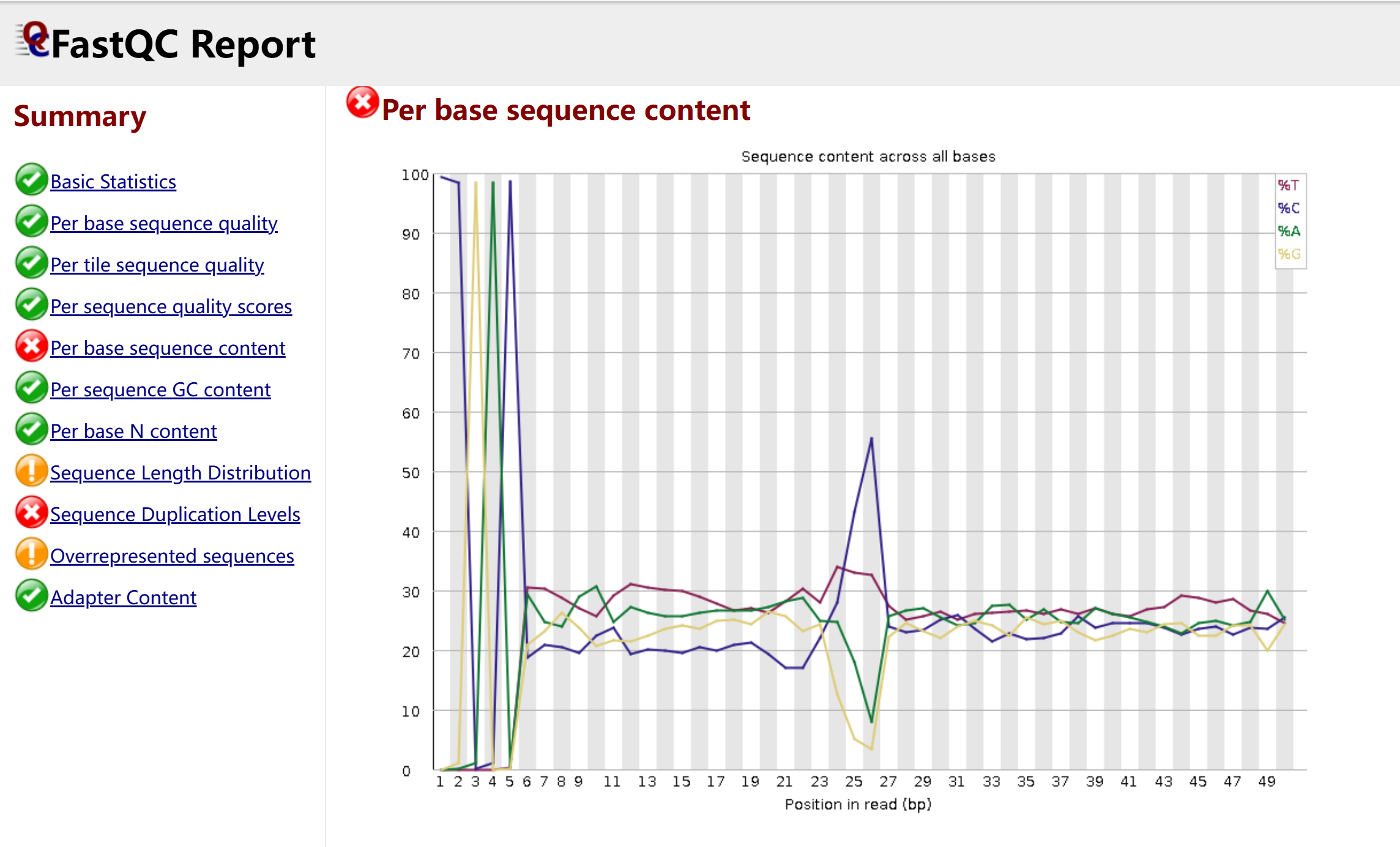
1:Per base sequence content(碱基组成异常)
原因(small RNA-seq 特有,无需担心)
-
miRNA 生物学特性:miRNA 5' 端有强的序列偏好性(通常第一个碱基是 U),会导致测序起始位点的碱基组成严重偏离随机分布(A/T/C/G 各 25%)。
-
接头残留信号:虽然接头序列已被切除,但 small RNA 接头的短片段残留(如 1-3 个碱基)仍会干扰起始位点的碱基组成。
-
非编码 RNA 保守区:rRNA、tRNA 等污染序列的保守区也会造成特定位置的碱基组成失衡。
应对方案
-
无需修正:这是 small RNA-seq 的正常生物学现象,不会影响后续的比对、定量分析。
-
过滤污染后会改善:当你用之前的水稻污染库(rRNA/tRNA/snRNA/snoRNA)去除污染序列后,这个指标会显著好转。
2:Sequence Duplication Levels(序列重复率极高)
原因(small RNA-seq 核心特征,完全正常)
-
miRNA 高丰度表达:少数 miRNA(如看家 miRNA)在细胞中表达量极高,测序后会产生大量完全相同的序列,导致重复率飙升(你的数据去重后仅剩余 5.63%,是 small RNA-seq 的典型数值)。
-
PCR 扩增偏好性:small RNA 文库构建需要 PCR 扩增,高丰度 miRNA 会被优先扩增,进一步提高重复率。
应对方案
-
下游分析分情况处理:
-
miRNA 定量:必须保留重复序列,因为重复数代表表达量,去重会导致定量完全错误。
-
新 miRNA 预测:可以适当去重,减少计算量。
-
-
红叉不代表数据质量差:对于 small RNA-seq,重复率红叉是合格数据的标志(相反,重复率低反而可能说明 miRNA 表达量低或数据有问题)。
3、去除冗余和非目标 RNA 的 reads
建库的 polyA 富集步骤已天然剔除了无 polyA 尾的 ncRNA,数据背景干净
-
Oligo(dT) 磁珠富集 polyA mRNA
-
去除 rRNA
-
目标是 miRNA 靶标预测(降解组)
这就是标准植物 PARE / degradome 思路。
不需要再额外去除 ncRNA
原因很简单:
-
polyA 富集本身已经排除了:
-
rRNA
-
tRNA
-
snRNA
-
大部分非编码 RNA
-
-
miRNA 靶标分析只关心:
-
mRNA 上的切割位点
-
再去 Rfam 属于重复操作。
而且:
-
你是植物
-
大部分公开文章都不做额外 ncRNA 去除
-
CleaveLand / PAREsnip 标准流程也是直接 transcriptome
所以这里不用再折腾。
场景:
-
无参 Trinity 拼接
-
植物降解组(polyA 富集)
-
目标是做 miRNA 靶标切割验证
-
不是研究可变剪接
用 unigene.fa 作为 degradome 比对参考。
为什么是 unigene,而不是 Trinity?
你要理解 degradome 的核心统计逻辑:
Category 0–4 是在“一个转录本内部”做 5' cleavage abundance 排名
如果你用 Trinity.fa:
-
一个基因拆成多个 isoform
-
reads 被分散到多个转录本
-
cleavage peak 被稀释
-
Category 可能下降
-
假阴性概率增加
在无参拼接情况下:
-
isoform 可能是拼接碎片
-
3'UTR 不完整
-
冗余严重
这会严重影响 degradome peak 判定。
而 unigene.fa 做了什么?
corset 聚类去冗余,每基因保留最长转录本
这等于:
-
collapse 到 gene-level
-
去除 isoform 冗余
-
集中 cleavage reads
-
peak 更清晰
-
Category 更稳定
这更适合 degradome 的统计模型。
什么时候才用 Trinity?
只有当你研究的是:
-
isoform-specific miRNA regulation
-
可变剪接调控
-
3'UTR 差异结合
否则没必要增加噪音。
构建转录本索引并比对降解组read(×)和下一步骤重复
1. 建bowtie2索引
bowtie2-build transcripts.fasta transcripts
2. 比对(-k 1 只保留最优比对;-L 15 种子长度)
bowtie2 -x transcripts -U degradome_clean.fq -k 1 -L 15 -p 8 -S deg.sam
3. SAM→BAM→排序→建索引
samtools view -bS deg.sam | samtools sort -@8 -o deg_sorted.bam samtools index deg_sorted.bam
#!/bin/bash
# 功能:批量将cutadapt过滤后的fq文件比对到无参转录本,并输出标准排序BAM
# 适配:降解组(PARE) / sRNA 比对到 Trinity 组装的 unigene/transcripts
# 输入:*_cutadapt.fq
# 输出:*.sam (标准格式)、*_sorted.bam、*.bai
# ===================== 核心配置区(请务必修改这里)=====================
REFERENCE="./unigene.fasta" # 你的无参转录本参考序列(绝对路径或相对路径)
THREADS=8 # 线程数,根据服务器配置调整
#ENV_NAME="trinity" # 你的mamba环境名(需包含bowtie2和samtools)
# =======================================================================
# 自动推导索引前缀(与参考序列同名)
INDEX_PREFIX="unigene"
echo -e "===== 降解组批量比对流程开始 ====="
echo -e "参考序列: ${REFERENCE}"
echo -e "索引前缀: ${INDEX_PREFIX}"
echo -e "线程数: ${THREADS}"
echo -e "使用环境: ${ENV_NAME}"
echo -e "=====================================\n"
# 第一步:检查索引是否存在(仅检查,不构建)
if [ ! -f "${INDEX_PREFIX}.1.bt2" ]; then
echo -e "❌ 错误:未检测到Bowtie2索引文件!"
echo -e "请先手动运行以下命令构建索引:"
echo -e "mamba run -n bowtie bowtie2-build ${REFERENCE} ${INDEX_PREFIX}"
exit 1
else
echo -e "✅ 检测到已有索引,直接开始比对。\n"
fi
# 第二步:批量查找并比对fq文件
find ./ -type f -name "*_cutadapt.fq" | while read CLEAN_FQ; do
# 1. 定义输出文件名
OUT_DIR=$(dirname "${CLEAN_FQ}")
BASE_NAME=$(basename "${CLEAN_FQ}" "_cutadapt.fq")
SAM_FILE="${OUT_DIR}/${BASE_NAME}.sam"
SORTED_BAM="${OUT_DIR}/${BASE_NAME}_sorted.bam"
echo -e "-------------------------------------"
echo -e "正在处理样本: ${BASE_NAME}"
echo -e "输入数据: ${CLEAN_FQ}"
echo -e "输出SAM: ${SAM_FILE}"
echo -e "输出BAM: ${SORTED_BAM}"
# 2. 执行标准Bowtie2比对(核心步骤:生成带头部的标准SAM)
# 参数说明:-k 1 仅保留最佳比对(降解组分析常用),-q 输入为fq格式
mamba run -n bowtie bowtie2 \
-x ${INDEX_PREFIX} \
-U ${CLEAN_FQ} \
-S ${SAM_FILE} \
-p ${THREADS} \
-k 1 \
-q
if [ $? -ne 0 ]; then
echo -e "❌ 比对失败: ${BASE_NAME}"
continue
fi
# 3. 转换为排序BAM并建索引(这次一定能成功,因为SAM是标准的)
echo -e "正在转换为BAM并排序..."
mamba run -n samtools samtools view -bS ${SAM_FILE} | \
mamba run -n samtools samtools sort -@ ${THREADS} -o ${SORTED_BAM}
# 4. 建立索引
mamba run -n samtools samtools index ${SORTED_BAM}
# 5. 结果检查
if [ -f "${SORTED_BAM}" ] && [ -f "${SORTED_BAM}.bai" ]; then
echo -e "✅ 全部完成: ${BASE_NAME}"
# 可选:删除中间SAM文件节省空间(如需保留请注释下行)
# rm ${SAM_FILE}
else
echo -e "❌ BAM处理失败: ${BASE_NAME}"
fi
done
echo -e "\n====================================="
echo -e "📌 批量比对任务全部执行完毕!"
echo -e "✅ 结果文件:*_sorted.bam 和 *.bai"
echo -e "✅ 这些文件可用于IGV可视化和后续深度分析。"
提取5’端切割位点(降解组信号)
1.# 用bedtools生成降解组密度文件(5’端位置计数)
bedtools genomecov -ibam deg_sorted.bam -5 -d > degradome_coverage.txt
2.# 格式转换为CleaveLand4输入(*.deg) # 格式:转录本ID 位置 深度
awk '{print $1"\t"$2"\t"$3}' degradome_coverage.txt > degradome.deg
#!/bin/bash
#=============================
# 功能:批量将 *_sorted.bam 生成降解组密度文件 (*.deg)
# 适配:CleaveLand4 降解组分析
# 输入:*_sorted.bam
# 输出:*.deg(转录本ID 位置 深度)
#=============================
# ===================== 核心配置区(请务必修改这里)=====================
THREADS=8 # 线程数,根据服务器配置调整
ENV_NAME="bedtools" # 你的mamba环境名(需包含bedtools)
# =======================================================================
echo -e "===== 降解组密度文件批量生成流程开始 ====="
echo -e "线程数: ${THREADS}"
echo -e "使用环境: ${ENV_NAME}"
echo -e "=============================================\n"
# 第二步:批量查找并处理BAM文件
find ./ -type f -name "*_sorted.bam" | while read SORTED_BAM; do
# 1. 定义输出文件名
OUT_DIR=$(dirname "${SORTED_BAM}")
BASE_NAME=$(basename "${SORTED_BAM}" "_sorted.bam")
COV_FILE="${OUT_DIR}/${BASE_NAME}_coverage.txt"
DEG_FILE="${OUT_DIR}/${BASE_NAME}.deg"
echo -e "-------------------------------------"
echo -e "正在处理样本: ${BASE_NAME}"
echo -e "输入BAM: ${SORTED_BAM}"
echo -e "输出coverage: ${COV_FILE}"
echo -e "输出deg: ${DEG_FILE}"
# 2. 执行bedtools统计5'端覆盖度(核心步骤)
echo -e "正在计算5'端覆盖度..."
mamba run -n ${ENV_NAME} bedtools genomecov -ibam ${SORTED_BAM} -5 -d > ${COV_FILE}
if [ $? -ne 0 ]; then
echo -e "❌ bedtools 失败: ${BASE_NAME}"
continue
fi
# 3. 转换为CleaveLand4标准格式
echo -e "正在转换为 *.deg 格式..."
awk '{print $1"\t"$2"\t"$3}' ${COV_FILE} > ${DEG_FILE}
# 4. 结果检查
if [ -f "${DEG_FILE}" ]; then
echo -e "✅ 全部完成: ${BASE_NAME}"
# 可选:删除中间coverage文件节省空间(如需保留请注释下行)
# rm ${COV_FILE}
else
echo -e "❌ deg 文件生成失败: ${BASE_NAME}"
fi
done
echo -e "\n============================================="
echo -e "📌 批量降解组密度文件生成任务全部执行完毕!"
echo -e "✅ 结果文件:*.deg"
echo -e "✅ 这些文件可直接用于CleaveLand4降解组分析。"
三、miRNA靶标预测与降解组验证(核心分析)
1.准备miRNA序列(miRNA.fasta)
| 文件 | 全称 | 序列类型 | 长度 | 主要用途 |
|---|---|---|---|---|
| mature.fa | Mature miRNA sequences | 成熟 miRNA | 20–23 nt | 用于 reads 比对、定量、差异分析 |
| hairpin.fa | Hairpin precursor sequences | miRNA 前体 | ~60–80 nt | 用于预测新 miRNA、分析前体结构 |
2.用CleaveLand4做降解组靶标鉴定
#!/bin/bash
#=============================
# 功能:批量运行CleaveLand4降解组分析
# 适配:降解组(PARE) + miRNA靶基因预测
# 输入:*.deg(降解组密度文件)
# 输出:每个样本独立的cleaveland_out目录
#=============================
# ===================== 核心配置区(请务必修改这里)=====================
MIRNA_FILE="./osa_mature.fa" # miRNA序列文件(绝对/相对路径)
TRANSCRIPT_FILE="./unigene.fasta" # 转录本序列文件(与bowtie2比对时一致)
THREADS=8 # 线程数
# =======================================================================
echo -e "===== 批量CleaveLand4降解组分析流程开始 ====="
echo -e "miRNA文件: ${MIRNA_FILE}"
echo -e "转录本文件: ${TRANSCRIPT_FILE}"
echo -e "线程数: ${THREADS}"
echo -e "=============================================\n"
# 检查输入文件是否存在
if [ ! -f "${MIRNA_FILE}" ]; then
echo -e "❌ 错误:miRNA文件不存在: ${MIRNA_FILE}"
exit 1
fi
if [ ! -f "${TRANSCRIPT_FILE}" ]; then
echo -e "❌ 错误:转录本文件不存在: ${TRANSCRIPT_FILE}"
exit 1
fi
# 第二步:批量查找并处理所有.deg文件
find ./ -type f -name "*.deg" | while read DEG_FILE; do
# 1. 定义输出目录(与deg文件同目录,以样本名命名)
OUT_DIR=$(dirname "${DEG_FILE}")
BASE_NAME=$(basename "${DEG_FILE}" ".deg")
CLEAVELAND_OUT="${OUT_DIR}/cleaveland_${BASE_NAME}"
echo -e "-------------------------------------"
echo -e "正在处理样本: ${BASE_NAME}"
echo -e "输入降解组文件: ${DEG_FILE}"
echo -e "输出目录: ${CLEAVELAND_OUT}"
# 2. 执行CleaveLand4(核心步骤)
echo -e "正在运行CleaveLand4..."
mamba run -n cleaveland4 CleaveLand4.pl -d ${DEG_FILE} -u ${MIRNA_FILE} -n ${TRANSCRIPT_FILE} -o ${CLEAVELAND_OUT}
if [ $? -eq 0 ]; then
echo -e "✅ CleaveLand4 分析完成: ${BASE_NAME}"
echo -e "✅ 结果目录: ${CLEAVELAND_OUT}"
else
echo -e "❌ CleaveLand4 分析失败: ${BASE_NAME}"
fi
done
echo -e "\n============================================="
echo -e "📌 批量CleaveLand4分析任务全部执行完毕!"
echo -e "✅ 每个样本的结果保存在 cleaveland_样本名 目录中"
echo -e "✅ 可在对应目录中查看靶基因预测结果和统计报告。"
关键参数说明(修正后)
- -m 1 :强制指定模式 1(对齐降解组 + miRNA + 转录本,最常用)。
- -d :降解组文件(必须有)。
- -u :miRNA 序列文件(替代原来的 -p ,模式 1 要求用 -u )。
- -t :转录本序列文件。
- -o :输出目录。
步骤:
raw fq
↓
cutadapt
↓
clean fq
↓
CleaveLand4 Mode 1
↓
自动完成:
- 建 bowtie index
- 比对
- 生成 density
- GSTAr
- 统计
- T-plot
把 fq 转成 fa:
find ./ -type f -name "*_cutadapt.fq" | while read file; do
out=${file%.fq}.fa
echo "Converting $file -> $out"
seqtk seq -a $file > $out
done转换原理:
FASTQ:
@read1
ACTG
+
IIII
转成 FASTA:
>read1
ACTG
CleaveLand4 只需要序列,不需要质量值。
#!/bin/bash
# ==========================
# CleaveLand4 批量 Mode 1 运行脚本
# ==========================
MIRNA_FILE="./single.fa"
TRANSCRIPT_FILE="./unigene.fasta"
THREADS=8
echo "===== CleaveLand4 Mode 1 批量运行开始 ====="
echo "miRNA文件: ${MIRNA_FILE}"
echo "转录本文件: ${TRANSCRIPT_FILE}"
echo "线程数: ${THREADS}"
echo "==========================================="
# 检查文件
if [ ! -f "${MIRNA_FILE}" ]; then
echo "❌ miRNA 文件不存在"
exit 1
fi
if [ ! -f "${TRANSCRIPT_FILE}" ]; then
echo "❌ 转录本文件不存在"
exit 1
fi
# 查找所有 cutadapt 后的 fq 文件
find ./ -type f -name "*_cutadapt.fa" | while read DEG_READS; do
SAMPLE_DIR=$(dirname "${DEG_READS}")
SAMPLE_NAME=$(basename "${SAMPLE_DIR}")
OUT_DIR="${SAMPLE_DIR}/cleaveland_${SAMPLE_NAME}"
echo "---------------------------------------"
echo "正在处理样本: ${SAMPLE_NAME}"
echo "输入文件: ${DEG_READS}"
echo "输出目录: ${OUT_DIR}"
echo "---------------------------------------"
mamba run -n cleaveland4 CleaveLand4.pl \
-e ${DEG_READS} \
-u ${MIRNA_FILE} \
-n ${TRANSCRIPT_FILE} \
-o ${OUT_DIR}
if [ $? -eq 0 ]; then
echo "✅ ${SAMPLE_NAME} 运行完成"
else
echo "❌ ${SAMPLE_NAME} 运行失败"
fi
done
echo "==========================================="
echo "🎯 全部样本运行完成"鉴定结果为空,问题排查:
# 查看单个文件大小(比如你的deg文件)
ls -lh 159-1_with_header.deg#查看是否进程在运行
ps aux | grep fq-fa.sh#随机抽 10 条 degradome read 看长度
head -20 159-2/159-2_cutadapt.fa
#统计 read 长度分布
awk 'NR%2==0{print length($0)}' 159-2_cutadapt.fa | sort | uniq -c
目标流程:处理_cutadapt.fa文件
-
去掉 5' 固定 5 nt
-
只保留 20–21 nt
-
输出最终 clean.fa
-
自动统计 reads 数量
-
保留目录结构
#!/bin/bash # ============================== # 批量处理 degradome fasta 文件 # 处理内容: # 1. 去掉 5' 前 5 nt # 2. 只保留 20–21 nt # 3. 输出 *_final.fa # ============================== # 遍历所有 cutadapt.fa 文件 find . -name "*_cutadapt.fa" -type f | while read file do echo "正在处理: $file" # 定义输出文件名 trim5_file="${file%.fa}_trim5.fa" final_file="${file%.fa}_final.fa" # Step 1️⃣ 去掉 5' 前 5 nt awk 'NR%2==1{print $0} NR%2==0{print substr($0,6)}' \ "$file" > "$trim5_file" # Step 2️⃣ 只保留 20–21 nt awk 'NR%2==1{header=$0} NR%2==0 && length($0)>=20 && length($0)<=21{ print header print $0 }' \ "$trim5_file" > "$final_file" # Step 3️⃣ 统计 reads 数量 count=$(grep -c ">" "$final_file") echo "最终 reads 数量: $count" echo "生成文件: $final_file" echo "-----------------------------------" done echo "全部处理完成。"你现在处理的是 FASTA 格式:
>read_id
SEQUENCE
>read_id
SEQUENCE不是 FASTQ。
FASTA 是:奇数行 header 偶数行 sequence 结构极其简单。
你现在做的操作是:固定裁剪前 5 nt 固定筛选 20–21 nt 这是纯文本长度操作,不涉及质量值、不涉及复杂解析。这种情况下,awk 非常安全。
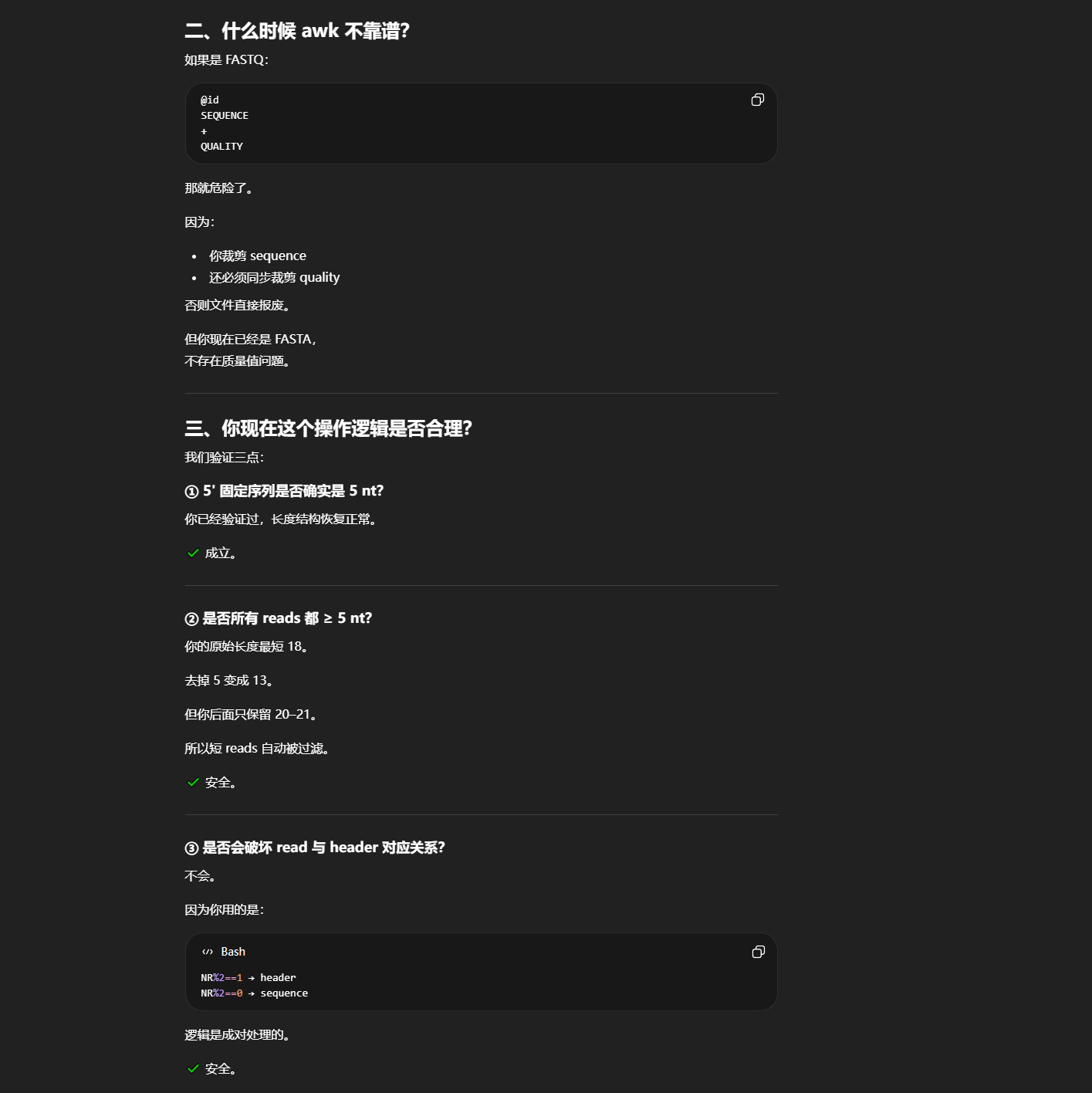
处理完后,随机抽一个样本再统计长度分布:
awk 'NR%2==0{print length($0)}' xxx_final.fa | sort | uniq -c
cutadapt.sh
#!/bin/bash
# ===================== 可配置参数 =====================
ROOT_DIR="./"
ADAPTER_SEQ_3="AGATCGGAAGAGCACACGTCTGAACTCCAGTCAC"
ADAPTER_SEQ_5="TCTTTCCCTACACGACGCTCTTCCGATCT"
POLYA_SEQ="A{10}"
QUAL_THRESH="20,20"
MIN_LEN=18
MAX_LEN=50
# ======================================================
echo "===== 开始批量执行cutadapt质控 ====="
echo "处理目录:${ROOT_DIR}"
echo "3'接头:${ADAPTER_SEQ_3}"
echo "5'接头:${ADAPTER_SEQ_5}"
echo "polyA过滤:${POLYA_SEQ}"
echo "质量阈值:${QUAL_THRESH}"
echo "长度范围:${MIN_LEN}-${MAX_LEN} nt"
echo "====================================="
# 激活cutadapt环境(比mamba run更稳)
source /mnt/software/miniforge3/etc/profile.d/conda.sh
conda activate cutadapt
# 查找所有未处理的fq文件
find ${ROOT_DIR} -type f -name "*.fq" ! -name "*_cutadapt.fq" | while read raw_fq; do
file_dir=$(dirname "${raw_fq}")
file_name=$(basename "${raw_fq}")
trimmed_fq="${file_dir}/${file_name%.fq}_cutadapt.fq"
echo "-------------------------------------"
echo "正在处理:${raw_fq}"
echo "输出文件:${trimmed_fq}"
cutadapt \
-g "${ADAPTER_SEQ_5}" \
-a "${ADAPTER_SEQ_3}" \
-a "${POLYA_SEQ}" \
-q "${QUAL_THRESH}" \
-m "${MIN_LEN}" \
-M "${MAX_LEN}" \
--trim-n \
-o "${trimmed_fq}" \
"${raw_fq}"
if [ $? -eq 0 ]; then
echo "✅ 处理成功:${file_name}"
else
echo "❌ 处理失败:${file_name}"
fi
done
echo "====================================="
echo "📌 批量cutadapt质控任务执行完毕!"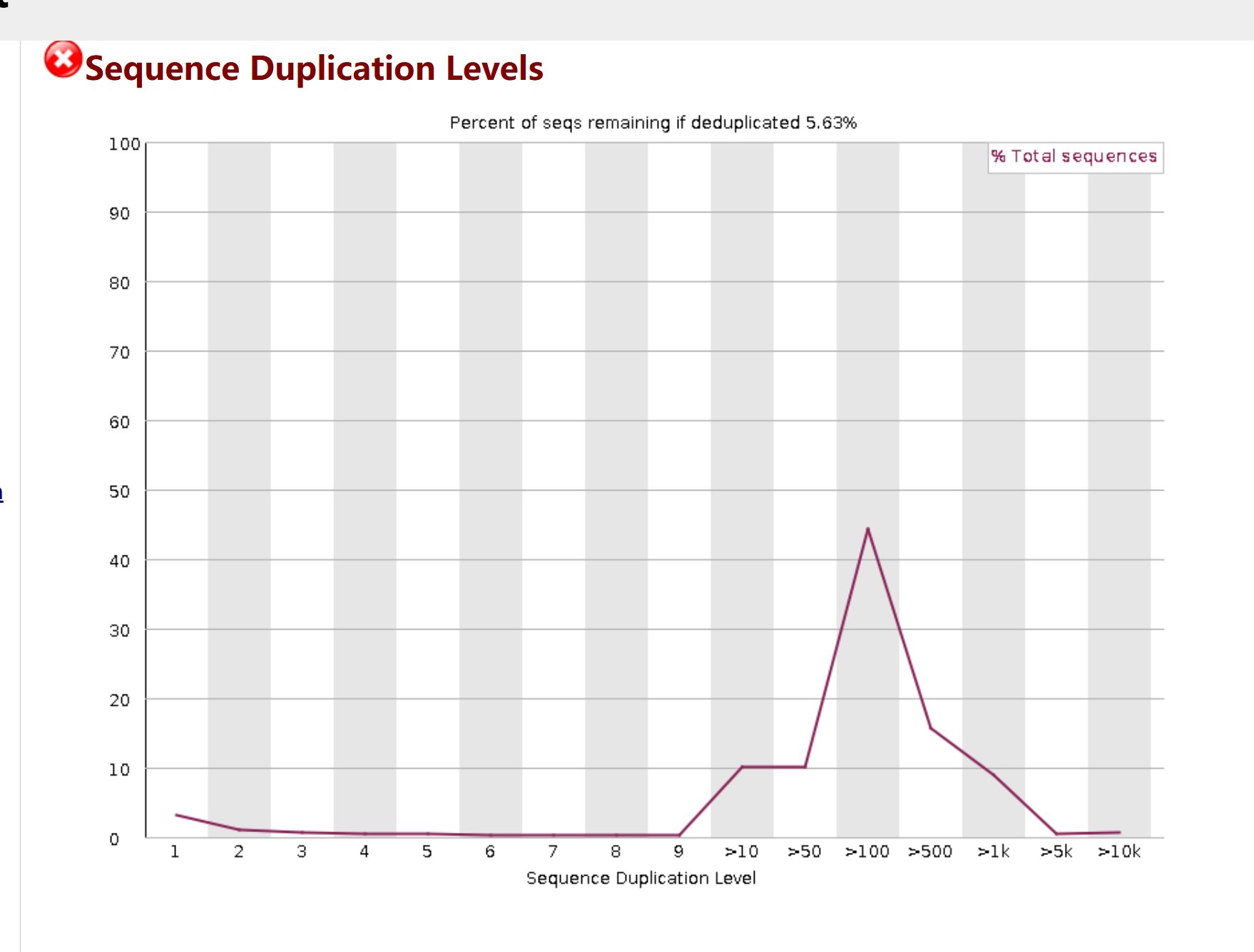

# 备份原始文件
cp IRGSP-1.0_transcript_2025-03-19.fasta IRGSP-1.0_transcript_2025-03-19.fasta.bak
# 1. 只保留 > 后面到第一个空格前的 ID,删除空格及后面所有内容
sed -i '/^>/ s/ .*//' IRGSP-1.0_transcript_2025-03-19.fasta
# 2. 清理掉可能残留的斜杠 / 等特殊字符
sed -i 's/\///g' IRGSP-1.0_transcript_2025-03-19.fasta如何查看某个目标基因
-
打开
*.cleavage.txt(一般在OUT_DIR根目录或者cleavage/文件夹里)。 -
用
grep筛选你的基因:
grep "LOC_Os01g01010" *.cleavage.txt-
LOC_Os01g01010替换成你感兴趣的基因 ID。 -
输出的列通常包括:
-
Transcript ID
-
miRNA ID
-
切割位置(nucleotide position)
-
read 数量(表示降解证据强度)
-
为什么默认没有显示已知靶基因
CleaveLand4 默认:
-
只显示在你的测序样本中检测到降解证据的转录本。
-
如果某个已知靶基因在样本中 没有降解 read 对应位置,就不会出现在
cleavage.txt或 T-PLOT 图里。 -
解决办法:
-
如果想强制检查已知靶标,即使没有 read,也可以用
-t target_genes.txt指定你的基因列表,CleaveLand4 会生成 T-PLOT 图(可能为空),然后你就知道该基因在样本中是否有降解证据。
-
grep -c "^@ID:" 159-1_cutadapt2.fa_dd.txt
腾讯云面向开发者汇聚海量精品云计算使用和开发经验,营造开放的云计算技术生态圈。
更多推荐
 18
18 0
0- 0



 已为社区贡献1条内容
已为社区贡献1条内容

所有评论(0)